
Abstract
As one of the most successful therapeutic target families, G protein-coupled receptors (GPCRs) have experienced a transformation from random ligand screening to knowledge-driven drug design. We are eye-witnessing tremendous progresses made recently in the understanding of their structure–function relationships that facilitated drug development at an unprecedented pace. This article intends to provide a comprehensive overview of this important field to a broader readership that shares some common interests in drug discovery.
Similar content being viewed by others


Introduction
G protein-coupled receptors (GPCRs) represent the largest protein family encoded by the human genome. Located on the cell membrane, they transduce extracellular signals into key physiological effects.1 Their endogenous ligands include odors, hormones, neurotransmitters, chemokines, etc., varying from photons, amines, carbohydrates, lipids, peptides to proteins. GPCRs have been implicated in a large number of diseases, such as type 2 diabetes mellitus (T2DM), obesity, depression, cancer, Alzheimer’s disease, and many others.2 Activated by external signals through coupling to different G proteins or arrestins, GPCRs elicit cyclic adenosine 3,5-monophosphate (cAMP) response, calcium mobilization, or phosphorylation of extracellular regulated protein kinases 1/2 (pERK1/2).3 The seven-transmembrane protein property endows them easy to access, while the diversified downstream signaling pathways make them attractive for drug development.4 The human GPCR family is divided into classes A (rhodopsin), B (secretin and adhesion), C (glutamate), and F (Frizzled) subfamilies according to their amino acid sequences (Fig. 1). Of the 826 human GPCRs, approximately 350 non-olfactory members are regarded as druggable and 165 of them are validated drug targets (Fig. 1 and Table S1).4,5,6 Latest statistical data indicate that 527 Food and Drug Administration (FDA)-approved drugs4 and ∼60 drug candidates currently in clinical trials target GPCRs (Table S1).5

Phylogenetic tree of GPCRs as drug targets. Node represents GPCR named according to its gene name. Receptors with approved drugs on the market are highlighted by color. GPCRs are organized according to GPCR database.4 Approved drug list was derived from previous publications,4,11 complemented by additional search of newly approved entities at Drugs@FDA (accessdata.fda.gov) until June 2020. See Table S2 for details
Started with crystal structure determination and accelerated by cryo-electron microscopy (cryo-EM) technology, three-dimensional (3D) structural studies on a variety of GPCRs in complex with ligands, G proteins/arrestins, or both7,8,9,10 (involving 455 structures from 82 different receptors) significantly deepened our knowledge of molecular mechanisms of signal transduction. Novel insights into ligand recognition and receptor activation are gained from inactive, transitional, active, and apo states, thereby offering new opportunities for structure-based drug design (SBDD).11 Pharmacological parameters such as cAMP accumulation, calcium flux, ERK phosphorylation, arrestin recruitment, and G protein interaction,12,13 are commonly used to evaluate ligand action and biased signaling. Ligand-binding kinetics and signaling timing render another dimension for interpreting signal bias profiles and link in vitro bioactivities with in vivo effects.14 In this process, a series of biased and allosteric modulators were discovered by rational design, ligand screening, and pharmacological assessment leading to the identification of novel binding sites or action modes.15,16
Apart from crystallography and cryo-EM, the striking advancement in GPCR biology is also attributable to the deployment of powerful technologies such as nuclear magnetic resonance (NMR), hydrogen–deuterium exchange (HDX), fluorescence resonance energy transfer, bioluminescence resonance energy transfer, surface plasmon resonance, single molecule fluorescence, CRISPR/Cas9, artificial intelligence, etc. This review systematically summarizes the latest information on this important drug target family to cover both basic and translational sciences in the context of drug discovery and development.
GPCR as drug target
Class A
Class A GPCRs, the so called “rhodopsin-like family” consisting of 719 members, are divided into several subgroups: aminergic, peptide, protein, lipid, melatonin, nucleotide, steroid, alicarboxylic acid, sensory, and orphan.17 They have a conventional transmembrane domain (TMD) that forms ligand-binding pocket and additional eight helices with a palmitoylated cysteine at the C terminal.18,19 Given the wide range of their physiological functions, this class of receptors is the most targeted therapeutically among all other classes. By manually curating Drugs@FDA original New Drug Application (NDA) and Biologic License Application (BLA) database (data extracted from August 2017 to June 2020) and cross-referencing with Drugbank,20 IUPHAR and ChemBL databases, we were able to find the approved drugs associated with this class.
Over 500 GPCR drugs target class A and many of them act at >1 receptor: 75% are made against aminergic receptors and 10% for peptidic ligand receptors with indications ranging from analgesics, allergies, cardiovascular diseases, hypertension, pulmonary diseases, depression, migraine, glaucoma, Parkinson’s disease to schizophrenia, cancer-related fatigue, etc. Approximately 500 novel drug candidates are in clinical trials. Of them, 134 are for peptide-activated GPCRs, while small molecules still occupy the majority. It is noted that 6% of class A members are sensory and alicarboxylic acid receptors that have broad untapped therapeutic potentials (Table S1). Chemokine, prostanoid and melanocortin receptors constitute >8% clinical trial targets in this class.
In the past 3 years, about 20 NDAs were approved targeting mostly peptide and aminergic receptors (Table 1). Siponimod and ozanimod provide alternatives to fingolimod (approved in 2010) for treating relapsing forms of multiple sclerosis by modulating sphingosine-1-phosphate receptor. Two radiolabeled ligands, gallium 68 dotatoc and lutetium 177 dotatate, have been approved for neuroendocrine tumor and pancreatic gastrointestinal cancer diagnosis, respectively. Pitolisant, a selective inverse agonist of histamine receptor, is used to treat narcolepsy-related daytime sleepiness, while lemborexant, an orexin receptor antagonist, is used for insomnia management. Gilteritinib (ASP2215) is a small molecule inhibitor of tyrosine kinase. However, it also antagonizes serotonin receptors without any reported pharmacological consequences. Revefenacin is a long-acting antagonist of muscarinic acetylcholine receptors (mAChRs) indicated for chronic obstructive pulmonary disease. Amisulpride, trialed for antiemetic and schizophrenia, was finally approved for antiemetic in 2020. This molecule is acting as an antagonist against dopamine and serotonin receptors. Fosnetupitant, a prodrug of netupitant, was approved for chemotherapy-induced nausea and vomiting. Cysteamine treats radiation sickness via modifying action of neuropeptide Y receptor. Cannabidiol is one the active constituents of the Cannabis plant and was trialed for schizophrenia, graft versus host disease, and anticonvulsant. It was eventually approved in 2018 for the treatment of severe forms of epilepsy—Lennox–Gastaut syndrome and Dravet syndrome. Meanwhile, fostamatinib, indicated for chronic immune thrombocytopenia, targets >300 receptors and enzymes, including adenosine receptor A3.
Class B
This class of GPCRs is divided into two subfamilies: secretin (B1) and adhesion (B2), containing 15 and 33 members, respectively.4,21 Secretin subfamily members are characteristic of large extracellular domains (ECDs) and bind to vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating peptide (PACAP), corticotropin-releasing factor (CRF), parathyroid peptide hormone (PTH), growth hormone-releasing hormone (GHRH), calcitonin gene-related peptide (CGRP), glucagon, and glucagon-like peptides (GLPs), respectively. Adhesion subfamily has nine subgroups, possessing unique N-terminal motifs, such as epidermal growth factor, cadherin, and immunoglobulin domains. They are distinguished from other GPCRs due to their roles in cell adhesion and migration.22,23 Apart from the long N-terminal domain, other unique features of the B2 subfamily are the GPCR autoproteolysis-inducing domain and the proteolysis site that are responsible for signaling activation through a Stachel sequence (a tethered agonist) and producing N-terminal fragment (NTF) and C-terminal fragment. The hallmarks of the B2 GPCR subfamily are a two-step activation model, the ligand–NTF interaction and the Stachel signaling/basal activity. Adhesion receptors can also signal independently of fragment dissociation and this has complicated pharmacological consequences.22,24,25
In this class, receptors of glucagon family peptides, followed by CGRP, PTH, GHRH, CRF, VIP, and PACAP, constitute major targets for therapeutic intervention (Table S1) of various diseases, including obesity, T2DM, osteoporosis, migraine, depression, and anxiety.26,27
To date, multiple GLP-1 receptor (GLP-1R) agonists have been developed by a combination of selective amino acid substitutions, enzymatic cleavage blockade, and conjugation to entities that increase binding to plasma proteins. These methods not only slow down fast renal clearance of the peptides but also extend their half-lives. Dose-dependent side effects such as nausea and gastrointestinal adverse events are the main drawbacks that are becoming more of a compliant with dose scaling.28,29 For instance, one newly approved GLP-1R agonist, semaglutide, has a noticeable half-life of 168 h thereby allowing weekly subcutaneous administration, while oral semaglutide (approved in 2019) formulated using absorption enhancer shows a similar half-life but is dosed daily with reported side effects (Table 2).30,31
One of the latest approaches to develop more efficacious therapeutics against T2DM and obesity relates to dual- and tri-agonists targeting two or more of GLP-1R, glucagon receptor (GCGR), and glucose-dependent insulinotropic peptide receptor (GIPR). Many of them are currently in different phases of clinical trials (Table 3).32,33,34,35,36,37 Of note, in this receptor family, GLP-2 stimulates intestinal growth and an approved GLP-2R agonist, teduglutide, is used to treat short bowel syndrome.38
CGRP family has a considerable clinical relevance. For instance, pramlintide that targets amylin receptor is utilized to treat both type 1 and type 2 diabetes. Salmon calcitonin has been explored as a treatment for Paget’s disease and metabolic disorders.39,40,41 Furthermore, the association of migraine and CGRP elevation led to FDA-approved monoclonal antibodies (mAbs) against its receptor, e.g., erenumab and eptinezumab, as well as several small molecule antagonists such as rimegepant and ubrogepant (Table 2).42,43 Two approved diagnostic agents are analogs of CRF (corticorelin ovine triflutate peptide) and GHRH (sermorelin) for diagnosis of Cushing’s disease or ectopic adrenocorticotropic hormone syndrome and growth hormone deficiency, respectively.44,45 Tesamorelin, another synthetic form of GHRH, was approved in 2010 to treat human immunodeficiency virus (HIV)-associated lipodystrophy.44
PTH analogs, teriparatide and abaloparatide, were approved in 2002 and 2017, respectively, for postmenopausal osteoporosis with similar side effects. However, abaloparatide binds to parathyroid hormone 1 receptor (PTH1R) with higher affinity and selectivity that resulted in greater bone density.46
No therapeutic agent from the adhesion subfamily has entered clinical trial to date (Table S1).2,4,47 Although, adhesion GPCRs have shown coupling to heterotrimeric G proteins, the major challenge associated with this family is connecting G protein signals with biological activities.24 This subfamily was found to play functional roles in the immune, cardiovascular, respiratory, nervous, musculoskeletal, reproductive, renal, integumentary, sensory, endocrine, and gastrointestinal systems, with implications in neurological and neoplastic disorders.24 For instance, ADGRG1 and ADGRF1 are considered as potential drug targets due to their extensive pathogenetic involvement. Two ADGRG1/ADGRG5 modulators, dihydromunduletone and 3-α-acetoxydihydrodeoxygedunin developed via drug screening efforts, showed disease-related efficacy changes thereby calling for exploration of their activities in a pathological environment.24,25 However, associated drug resistance may not only hamper disease but also offer insights into potential mechanisms of such resistance and strategies to tackle it.
Classes C and F
Class C (glutamate) contains 22 receptors, which are further divided into 5 subfamilies including 1 calcium-sensing receptor (CaSR), 2 gamma-aminobutyric acid (GABA) type B receptors (GABAB1 and GABAB2), 3 taste 1 receptors (TS1R1–3), 8 metabotropic glutamate receptors (mGluR1–8), and 8 orphan GPCRs.48 The distinctive features of glutamate subfamily are their large ECD and obligated constitutive dimer for receptor activation.49 The structural information of ECD indicates the roles of conserved venus fly trap (VFT) and cysteine-rich domain (CRD) on the ligand-binding site. Two conserved disulfide bonds between VFT domains stabilize the homodimers or heterodimers of class F GPCRs.50 The cryo-EM structures of the first full-length mGluR551 and more recently the GABABRs further revealed their assembly mechanism and overall architecture.52,53,54,55 To date, 16 drugs have been approved by the FDA targeting 8 class C GPCRs. As archetypal receptors, mGluRs mediate the stimulus of agonists such as glutamate and their malfunction are implicated in various diseases, including cancer, schizophrenia, depression, and movement disorders. Acamprosate, an antagonist of mGluR5, was launched in 2004 as an anti-neoplastic agent.56 In fact, mGluRs have been vigorously pursued as therapeutic targets and there are 15 drug candidates undergoing clinical trials at present for pain, migraine, Parkinson’s disease, Fragile X syndrome, etc. Although allosteric modulators of class C have attracted significant development efforts involving 8 clinical trial stage compounds [2 positive (PAM) and 6 negative (NAM) allosteric modulators], the only success is cinacalcet, a small molecule PAM of CaSR approved in 2004 for hyperparathyroidism and calcimimetics.57
Only one class F GPCR (smoothed receptor SMO) has been validated as a drug target whose small molecule antagonists were approved as anti-neoplastic agents.58 Other 10 members of this class are all Frizzled receptors (FZD1–10), which mediate Wnt signaling and are essential for embryonic development and adult organisms. FZDs together with cognate Hedgehog and Wnt signal are associated with a variety of diseases such as cancer, fibrosis, and neurodegeneration.59 They share a conserved CRD in the extracellular part and ECD structures of SMO and FZD2/4/5/7/8 were determined.60 However, only SMO, FZD4, and FZD5 have TMD structures.61,62,63 Lack of full-length structures and complexity in signaling pathways impeded drug discovery initiatives.60 Linking of Wnt with extracellular CRD would activate downstream signaling, while the dimerization process and the interaction between CRD and TMD remain elusive.64 It is known that the downstream effectors of Wnt signaling consist of β-catenin, planar cell polarity, and Ca2+ pathways, whereas receptor activation involves in Wnt, Norrin, FZD, LDL receptor-related protein 5/6, and many other co-factors.64 Key breakthrough is thus required to advance our knowledge of these receptors.
Medicinal chemistry of GPCR
Agent type
Agents targeting GPCRs continue to expand in the past decades. Among them, exogenous small molecules, including traditionally developed synthetic organics, natural products, and inorganics, still dominate with a total percentage of 64% (Fig. 2). Nevertheless, the proportion of small molecules declines since 2010. In addition to traditional ligand discovery, several new modalities appear, though currently at the stage of academic research. Covalent ligands, with the embedding of reactive moieties that can be covalently linked to receptors, significantly enhance the weak binding of unoptimized leads.65,66 Photoactive ligands, developed by the introduction of photo-responsive groups to drug candidates, bring a new interdisciplinary field, photopharmacology. Albeit in its infancy, it has already found in vivo applications.67,68

Analysis on agents targeting GPCRs. Distribution of molecule type (left) and action mode (right). Positive, PAM; Negative, NAM
In comparison, biologicals, such as peptides, antibodies, and metabolites, become more and more visible in the list. Particularly, the number of approved peptide drugs occupies approximately one third of the whole repertoire, with many more in different clinical stages as the pipeline41,69—most of them target classes A and B GPCRs. Naturally occurring peptides have been continually discovered from plants, animals, fungi, and bacteria. Although they act as efficient chemical messengers to modulate cellular functions, these peptides suffer from unfavorable pharmacokinetic and pharmacodynamics properties, such as very short plasma half-lives and low plasma protein binding. Therefore, chemical modifications are required to promote the membrane permeability, brain penetration, and oral bioavailability.70 Available strategies include peptide cyclization, N-methylation, palmitoylation, unnatural amino acid insertion, peptide–small molecule conjugation, and peptide self-assembly. By the way, developing peptidic agents may offer a new approach to de-orphanize certain orphan GPCRs.71
mAbs represent a promising alternative in GPCR drug discovery.72,73 Over small molecules, mAbs possess obvious advantages of improved specificity, affinity, and other pharmacological properties. Thus they are being developed against cancers, inflammation, and metabolic disorders. To date, three GPCR-targeting mAbs were approved (mogamulizumab, erenumab, and eptinezumab) while bi-specific antibodies, nanobodies, antibody–drug conjugates, and antibody–peptide conjugation are also in the development stage.
The emergence of many conceptually new molecular entities, such as RNA aptamer, provides not only powerful tool for biophysical study but also potential therapeutic candidates.74 Usually, aptamer has great molecular diversity and little immunogenicity.75 In addition, GPCRs are known to function by forming dimers (homodimers or heterodimers) and oligomers on the cell membrane.76 Therefore, strategies to induce receptor dimerization and/or oligomerization have received attention using scaffolds based on DNA (aptamer), small molecule, and physical stimuli.77
Structure–activity relationship (SAR)
Studies of SARs are critical to the identification of drug-like molecules, especially when the crystal or cryo-EM structure of a drug target is not available. Given that many 3D GPCR structures have been solved in the past decade, most approved drugs were discovered without relevant structural information. Two examples are reviewed below to show the importance of SAR analysis.
Orexin-1 and orexin-2 receptors (also known as hypocretin receptors, OX1R and OX2R) are class A GPCRs for which two endogenous peptide ligands were identified, orexin A and orexin B (also known as hypocretin 1 and hypocretin 2). The orexin signaling system plays a crucial role in regulating the sleep/wake cycle—both OX1R and OX2R are involved while the precise contribution of each has yet to be defined. Therefore, dual antagonists were developed as potential treatment for insomnia.78 Suvorexant (belsomra), the first-in-class dual orexin receptor antagonist, was launched in 2014.79 The second, lemborexant/E2006 (dayvigo) developed by Eisai, was approved by the FDA in 2020.80 It started from hit compound 1 (6, Fig. 3) with modest binding affinity to OX2R (Ki = 8.7 µM) and no affinity for OX1R.81 The first round of SAR studies revealed that changing the ketone group to an amide led to a remarkable enhancement (~1000-fold) of binding affinity at both OX1R and OX2R (compound 2). Substitution of the aniline group with a 2-amino-5-cyano pyridine (compound 3) maintained OX2R affinity and reduced OX1R activity, but physicochemical properties were improved compared to compound 2.81 Further SAR studies focused on the modification of all three aromatic substitutions in compound 3.82 Changing the di-OMe-phenyl substituent to a pyrimidine group resulted in a significant loss of binding affinity, as shown with compound 4, but an improved overall profile due to reduced lipophilicity and enhanced solubility. Then replacing the cyano group to a fluorine regained the binding affinity for both receptors (compound 5), and finally adding a second fluorine to the benzene group significantly improved OX1R affinity and led to lemborexant.82 Clearly, slight structural modifications may cause significant change of compound activity, and SAR studies coupled with optimization of physicochemical properties are useful steps to obtain druggable candidates.

SAR studies that led to the discovery of the dual orexin receptor antagonist lemborexant
CGRP is a 37-amino acid neuropeptide and its receptor is implicated in migraine.83 The benzodiazapinone compound 7 was identified as a hit compound with modest CGRP receptor binding affinity (Ki = 4.8 µM, Fig. 4).84 Replacing the right-hand spirohydantoin structure with piperidyldihydroquinazolinone, a privileged structure for CGRP receptor antagonists,85 an affinity boost of 100-fold was gained.84 Further optimization of the benzodiazepinone core resulted in the caprolactam compound 9, which showed a Ki of 25 nM.86 Changing the piperidyldihydroquinazolinone moiety to a piperidylazabenzimidazolone led to compound 10, with a binding affinity of 11 nM.87 Then by changing the N-substituent on the caprolactam and adding di-fluro substitutions on the lower benzene ring delivered compound MK-0974 (11, Ki = 0.77 nM, Fig. 4), which entered clinical trials. Compound 12 (BMS-846372) shares the same piperidylazabenzimidazolone and the lower diflurobenzene substructures with 11 but differs from the latter with a carbamate core structure and a pyridine-fused-cyclopentane in replacement of the caprolactam.88 Compound 11 displayed high binding affinity while suffered from poor physicochemical properties, such as low solubility. To improve this, a hydroxyl group was attached to the cycloheptane ring and it was discovered that the (S)-isomer 13 was more potent than the (R)-OH compound 14. The –OH was finally replaced with an -NH2 group, which led to the clinical compound rimegepant.89 The latter was further developed for better safety and efficacy profiles and obtained regulatory approval by the FDA in 2020.

SAR studies that resulted in the discovery of CGRP antagonists
The above examples demonstrate that, starting from a modest affinity hit compound, systematic SAR studies could successfully lead to very potent GPCR ligands that qualify as clinical candidates. Slight modifications of chemical structures sometimes cause remarkable changes of binding affinity or potency, which could not always be accurately predicted by conventional methods, such as docking. Therefore, SAR studies will continue to play a critical role in drug discovery.
GPCR structure
The structure of GPCRs is a crucial determinant for understanding the molecular mechanisms underlying ligand recognition and receptor activation. It provides a foundation for drug discovery. The first crystal structure of inactive state rhodopsin purified from bovine eyes was solved in 2000.90 Although tremendous efforts have been made, elucidation of GPCR structures remains challenging due to several bottlenecks, including low receptor expression level, difficulties in extraction, highly flexible conformation, lack of crystal contacts, etc. The first crystal structure of GPCR extracted from exogenously expressed host cells, the human β2-adrenergic receptor (β2AR, gene name: ADRB2) bound to an antagonist, was disclosed in 2007, representing a milestone in GPCR structural biology.7 Several innovative methods, especially the incorporation of a soluble fusion partner and lipidic cubic phase (LCP) crystallization, facilitated subsequent studies. Further technological breakthroughs in protein expression and purification,91,92 receptor engineering,8,93 application of Fab fragment and nanobody,94,95 and GPCR crystallization96 led to an exponential growth of this field.
The crystal structure of β2AR in complex with stimulatory G protein (Gs) solved in 201197 and rhodopsin bound to visual arrestin reported in 201598 revealed the molecular mechanism of GPCR interaction with G protein and arrestin, respectively. Notably, the wave of resolution revolution in the single-particle cryo-EM has brought a significant impact on the determination of GPCR complexes.10 Over 90% of GPCR–transducer complex structures were solved using cryo-EM (Table 4). To date, a total of 455 structures from 82 GPCRs belonging to all classes except B2 have been reported (Table 4). Although GPCRs show extensive sequence diversity, they share a conversed structural architecture of a TMD composed of seven helices embedded in the cell membrane. The transmembrane (TM) helices, essential for signal transduction across the cell membrane, are linked by three extracellular loops (ECLs) and three intracellular loops (ICLs). However, distinct structural features exist among members from different classes despite their overall structural similarity.
Various ligands of class A GPCRs bind to similar orthosteric sites directly in the helix bundle. Structural variations in ECLs, TM helices, and side chains show a remarkable variety of sizes, shapes, and physicochemical properties of the ligand-binding pockets, leading to diversified mechanisms of ligand recognition.99 For example, ligand binding, access, and selectivity are affected by ECL2.100,101 Many published GPCR structures are in an antagonist-bound inactive state, but the number of agonist-bound active state structures have been increased steadily in recent years due to the deployment of cryo-EM. Additionally, the structure of human M2R bound to a PAM (LY2119620) unveiled the allosteric ligand recognition mechanism.102 A summary of complicated recognition and modulation mechanisms of class A GPCRs bound to agonist, antagonist, and PAM is illustrated in Fig. 5a.

Structural features and common activation mechanism of class A GPCRs. a Ligand-binding pockets. Agonist, antagonist, and allosteric ligand are indicated as sticks in yellow, green, and salmon, respectively. Ligands are shown from the following structures (PDB code): 2RH1, 3PWH, 3VW7, 4IAR, 4MQT, 4PHU, 4RWS, 4XEE, 4XNV, 4Z35, and 4ZJ8. b–f The common activation pathway of class A GPCRs as exampled by the structures of inactive (gray, PDB code 3NYA) and active β2AR (green, PDB code 3SN6). The conformational changes of conserved “micro-switches”, including CWxP (b), PIF (c), Na+ pocket (d), NPxxY (e), and DRY (f), are highlighted. Side chains of residues in “micro-switches” are shown as sticks. Red arrows indicate the shift and swing directions of elements in the active β2AR structure relative to the inactive one
Class A GPCRs are activated through a common pathway, which strings the conserved “micro-switches” together, including CWxP, PIF, Na+ pocket, NPxxY, and DRY, thereby linking the ligand-binding pocket to the G protein-coupling region (Fig. 5b–f).99,103 The binding of diverse agonists triggers the rotameric switch of W6.48, a highly conserved residue in the “CWxP” motif, and the concomitant side chain rotation of F6.44 (Fig. 5b). Upon stimulation by an agonist, conformational rearrangement occurs in the PIF (P5.50, I3.40, and F6.44, Fig. 5c) and the Na+ pocket residues (D2.50, S3.39, N7.45, and N7.49, Fig. 5d). These reorganizations rigger the notable outward displacement of TM6, the hallmark of class A GPCR activation (Fig. 5b). The repacking of Na+ pocket residues initiates the TM7 movement toward TM3. Upon receptor activation, the “NPxxY” residue Y7.53 changes its rotamer conformation and points toward TM3, rendering new contact formation between Y7.53 and residues in TM3 (L3.43, I3.46, and R3.50, Fig. 5e) and subsequently the enhanced packing of TM3–TM7. Finally, “DRY”, one of the most conserved motifs in class A receptors, locates at the bottom of the 7TM and forms an intra-helical salt bridge between D/E3.49 and R3.50. R3.50 forms an additional inter-helical salt bridge with D6.30, known as the ionic lock, which connects the intracellular ends of TM3 and TM6 to stabilize receptors in an inactive state (Fig. 5f). These contacts are eliminated after agonist binding, and R3.50 is released to interact with other residues to facilitate the G protein coupling. It is notable that an acidic residue at position 6.30 is less conserved in 30% of class A receptors. Alternatively, R3.50 may form polar interactions with other polar residues in TM6 (i.e., T6.34 in κ-OR and μ-OR) to mediate the activation. Collectively, these rearrangements and reorganizations of conserved motifs are critical to the activation of class A GPCRs.
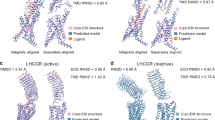
Class B GPCRs contain a large ECD and a TMD bundle with the peptide ligand recognition by both domains. According to the two-domain-binding model, the C-terminus of the peptide interacts with the ECD and orient the N-terminus of the peptide toward the TMD bundle. It then engages with the TMD core to facilitate receptor activation.104 The most remarkable structural feature of this class is the swing of ECD, accompanied by the corresponding shift of the peptide C-terminus (Fig. 6a, b). Conversely, the N-terminus inserts into a V-shape cavity within the helix bundle with a similar binding pose. Compared to small molecule-binding pocket of class A, that of class B is more solvent-accessible with higher flexibility and larger volume to accommodate sizeable peptidic ligands.9 In addition, structural studies also reveal an antagonist-binding pocket deep in the TMD bundle of CRF1R105 and a common binding site for allosteric modulators of GCGR106 and GLP-1R107 located outside the TMD bundle between TM6 and TM7 (Fig. 6c).

Structural features and common activation mechanism of class B GPCRs. a, b Structural features of the peptide-binding pocket. The shift of peptide C-terminus (a) and ECD (b) is indicated as red arrows. The peptides urocortin 1 (UCN1)1 bound to CRF1R (light blue, PDB code: 6PB0), UCN12 bound to CRF2R (salmon, PDB code: 6PB1), PACAP38 (red, PDB code: 6P9Y), long-acting PTH (LA-PTH, green, PDB code: 6NBF), GLP-1 (cyan, PDB code: 5VAI), sCT (yellow, PDB code: 6NIY), and CGRP (magenta, PDB code: 6PB1) are shown as cartoons. Binding poses of the antagonist (green) and allosteric ligand (salmon) are shown as sticks (c, PDB codes: 4K5Y, 5EE7, 4Z9G, 5VEW, and 5VEX). d, e The common activation mechanism of class B GPCRs as exampled by the structures of inactive GCGR (gray, PDB code 3NYA) and active VIP1R (green, PDB code 6VN7). Side chains of residues in three conserved polar network are shown in stick presentation. The conserved P6.47bxxG6.50b motifs in TM6 are shown as single red spheres
A comparison of the full-length active receptor structures with that in the inactive state reveals a general activation mechanism for class B GPCRs (Fig. 6d, e).9,108 The binding of a peptidic ligand causes a conformation rearrangement of the central polar network with simultaneous destabilization of the TM6 helix, thus initiating a sharp kink formation at the conserved motif P6.47bxxG6.50b. This central polar network is preserved across the class B receptors solved so far. However, the exact interactions may vary among different members in a ligand- and receptor-specific manner. The rearrangement of TM6 breaks the polar interaction of the conserved HETX motif and the TM2-6-7-helix 8 polar network, thereby inducing a notable outward displacement of TM6 and creating a cytoplasmic cavity to accommodate α5 helix of Gαs protein.
Class C GPCRs are distinguished by a characteristically large ECD that forms an obligate dimer. The ECD is distal to the TMD and contains an orthosteric ligand-binding pocket. It is composed of a ligand-binding VFT linked by the CRD to the TMD except for the metabotropic GABAB receptor (GABABR), which lacks CRD (Fig. 7a–d). This structural feature results in a potentially unique ligand recognition mechanism. The full-length structures of mGlu5 in apo and agonist-bound states,51 as well as several recently reported full-length structures of GABABRs,52,53,55 have significantly extended our understanding of the activation mechanism of the class C receptors. It is known that an agonist binds and stabilizes the conformation of the VFT, leading to compaction of the inter-subunit dimer interface and proximity of the CRD (Fig. 7a, b). This conformation transition, in turn, triggers TMD rearrangement through interaction between ECL2 and CRD.51,52,53,55 In contrast to mGlu5, the GABABR undergoes a featured asymmetric activation. After the binding of agonist baclofen to GABAB1 (GB1) subunit, the latter only exhibits a negligible conformational change. Additionally, due to lacking CRD in the GABABRs, the relatively shorter stalk and ECL2 region may rigidify their conformations and mediate the transduction of conformational changes from VFT to 7TM.53 In contrast, substantial conformational alterations occur at the stalk and TM3/4/5-ICL3 regions at the cytoplasmic part of GB2 (Fig. 7c, d), which predominantly couples to Gi1 heterotrimer. Interestingly, cholesterols are observed at the TMD interface of inactive GABABRs52 (Fig. 7c), while two chained phospholipids occupy a binding site overlapped with the orthosteric binding pocket in class A GPCRs52,53 (Fig. 7c, d). These cholesterols and phospholipids may contribute to the activity regulation of the GABABR. Noteworthy, in contrast to other allosteric modulators that bind to the TMD core of class C GPCRs (Fig. 7e),109,110,111 (+)-BHFF occupies a novel allosteric site at the interface of TMDs in GB1 and GB2 subunits52 (Fig. 7d). This novel allosteric binding site may provide a promising template for the design of PAMs for GABABRs.

Structural features and activation mechanism of class C GPCRs. The structures of mGlu5 in resting state (a, PDB code: 6N52) and active state (b, PDB code: 6N51), as well as GABABR in inactive (c, PDB code: 7C7S) and active states (d, PDB code: 7C7Q) are displayed, respectively. Agonists L-quisqualate (b, magenta) and antagonist CGP54626 (c, cyan) of mGlu5 as well as agonist baclofen (d, magenta) and allosteric modulator (+)-BHFF (d, yellow) of GABABR are shown as spheres. Cholesterols (c, yellow) and phospholipids (c, d, salmon) are indicated as sticks. Binding of allosteric ligands to TMD of class C GPCR is indicated as salmon sticks (e, PDB codes: 4OR2, 4OO9, 5CGC, and 6FFH)
Class F GPCRs include SMO and 10 FZDs in humans. Besides a canonical TMD across all classes of GPCRs, class F is characterized by a large ECD composed of a CRD and an ECD linker domain to connect with TMD (Fig. 8a, b).112 It was reported that SMO has a unique allosteric modulation mechanism.113 In fact, two ligand-binding sites have been identified: one in CRD and the other in TMD (Fig. 8b). SMO is activated by cholesterol via binding to CRD. The binding of an antagonist to TMD was proposed to trigger its conformation change thereby propagating to CRD and allosterically impeding the binding of cholesterol.113 Recent structural studies reveal that cholesterol and oxysterol that are critical for SMO activation are located deep within the 7TM domain of SMO (Fig. 8b, c).114,115 CRD of FZD can interact with lipoglycoprotein Wnt and Norrin (specific ligand for FZD4) to mediate the Wnt signaling.61,116 Structures of CRD in complex with Wnt or Norrin provided molecular details of how they formed a symmetrical homodimer (2:2 complex) during ligand recognition (Fig. 8d, e).117,118 In contrast to SMO, the ligand recognition and receptor activation mechanisms of FZD remains elusive due to the absence of the full-length FZD structures. So far, only two apo TMD structures of FZD4 and FZD5 have been reported (Fig. 8f).61,63 Structures of the full-length FZD in a ligand-bound state are required awaiting to provide mechanistic explanations.

Structural feature of class F GPCRs. a Superposition of SMO crystal structures bound to agonists (yellow sticks) and antagonists (green sticks). The following structures are shown (PDB codes): 4JKV, 4N4W, 4O9R, 4QIM, and 5V56. CRD and LD (linker domain) are highlighted; b A comparison of structures of full-length SMO in the active state (PDB codes: 5L7D and 6O3C). Cholesterols are indicated. SAG21K, the agonist of SMO, is shown as yellow spheres. c The cryo-EM structure of SMO TMD in complex with Gi heterotrimer (PDB code: 6OT0). The agonist 24(S),25-epoxycholesterol is shown as magenta sticks. d Crystal structures of the Wnt3-FZD8 CRD complex. e Crystal structures of the Norrin-FZD4 CRD complex. f A comparison of the apo TMD structures of FZD4 (PDB code: 6BD4, yellow) and FZD5 (PDB code: 6WW2, green)
GPCR pharmacology
The explosion of 3D GPCR structures and computational simulations has revealed the dynamic conformations between inactive, intermediate, and active states of GPCRs. The detailed structural information illustrated that cholesterol, ion, lipids, and water also participate in receptor activation.99,119 The flexibility of receptor-binding pocket endows the complex pharmacological mechanisms of ligand recognition and signal transduction. Biased signaling, allosteric modulation, and polypharmacology are helping us better understand how GPCRs bind to numerous ligands and how they transmit diverse signals to elicit physiological functions.
Polypharmacology
Ligand binding to multiple targets leads to antagonism, additive, or synergism pharmacological responses that could be positive or negative based on the mechanism of action. The paradigm of one drug vs. multiple targets has outpaced the time and cost associated with the conventional therapy.120 Polypharmacology thus emerges to study acceptable degree of specificity toward multiple targets, interconnected signaling pathways that result in clinical benefit or cross-reactivity that may cause adverse events.121,122 T2DM, obesity, cancer, and Alzheimer’s disease are major indications for GPCR modulators.4 These polygenic diseases are not completely treatable by a single agent, while desirable efficacies may be achieved for certain respiratory conditions, central nervous system (CNS) disorders, and cardiovascular diseases through modulators directed against β2AR, DRD2, and AGTR1,123 respectively.
It was shown that 5-hydroxytryptamine receptor 2 (5-HT2) binds to selective inverse (ritanserin) and highly promiscuous (ergotamine) agonists but the interaction with ergotamine is broad.121 This feature allows the development of pan serotonin receptor modulators to treat different diseases.124,125,126,127 For instance, zolmitriptan as an anti-migraine drug is also used for hyperesthesia via binding to off-target site,120 and lorcaserin (Belviq) is used to treat obesity while its therapeutic potential for depression, schizophrenia, and drug addiction is being investigated.128,129 However, off-target activity, hallucinations,130 and cardiac valvulopathy related to 5-HT2A and 5-HT2B modulation129 should be carefully monitored. Atypical antipsychotics are mainly targeting both dopamine and serotonin receptors, usually as antagonist for DRD2 and antagonist or inverse agonist for 5-HT2A.131 Exemplified by clozapine120 and aripiprazole,132 haloperidol, amoxapine, and asenapine4 display a diverse spectrum of receptor interaction. Additionally, carazolol, a member of aminergic division exerts its effects by interacting with multiple adrenergic receptors as inverse agonist or allosteric antagonist.19,129 Istradefylline combined with L-DOPA/dopamine simultaneously target A2AR, DRD1 and DRD2 in animal model of Parkinson’s disease.131 Amitryptyline, a tricyclic compound targeting muscarinic and histamine H1 receptors,133 is used to treat depression and non-selective muscarinic receptor antagonists are trialed for bladder dysfunction.4 Lorazepam, indicated for anxiety due to interaction with GABAAR, is also an allosteric modulator of the proton-sensing GPCR (GPR68)134 and has been repurposed to treat pancreatic cancer.2 6’-Guanidinonaltrindole (6’-GNTI) is an agonist with higher selectivity for δ/κ-opioid receptor heterodimer but not homodimer. Importantly, 6’-GNTI is an analgesic that offers additional benefit. In cardiovascular diseases, β blockers decrease catecholamine-induced heart rate elevation via interaction with valsartan (AT1R-mediated signaling).135 It is of note that mono-, dual-, and tri-agonists for the glucagon family of receptors (GLP-1R, GCGR, and GIPR) have been developed and trialed for weight loss and glucose control (Table 3). Successful outcome will determine whether unimolecular polypharmacology is a practical approach to translate safety and efficacy of multiple agents into a single molecule.136
Biased agonism
Activated GPCRs can recruit multiple transducers (such as heterotrimeric G proteins, GPCR kinases, and β-arrestin) and consequently produce distinct biological responses. Ligands that preferentially engage one signaling pathway over others are regarded as bias and may show improved therapeutic outcomes.137,138 Biased signaling that has been applied to drug discovery involve AT2R, µ-OR, κ-OR, β-adrenergic receptors, DRD2, CTR, CCR, and adenosine receptors. µ-OR is the best studied receptor for biased agonism.137 Compounds that stimulate Gαi coupling and cAMP production but not β-arrestin recruitment are preferable to retain analgesia and reduce opioid-related side effects.139 This G protein bias was also demonstrated with widely used drug tramadol,140 whose active metabolite, desmetramadol, elicited maximum cAMP production without affecting β-arrestin 2 recruitment compared to fentanyl and morphine. Safety profile is improved with less adverse effect such as respiratory depression.140 Another µ-OR-biased ligand, oliceridine (TRV130, OlinvoTM), passed phase III clinical trial but did not get the FDA approval for safety concerns.141 The NDA for oliceridine was resubmitted and a new counterpart, TRV734, is not only suitable for oral administration but also safer due to reduced dependency.142 A fourth µ-OR-biased ligand, PZM21, cross-reacts with κ-OR and failed to reduce respiratory depression in C57BL and CD-1 mice.143 Whether this relates to its residual but marked effect on β-arrestin 2 recruitment, as opposed to oliceridine whose action is negligible,144 remains to be further studied.
Similar situation occurred with κ-OR as well whose agonists possess analgesic property and have a low risk of dependence and abuse but with adverse effects such as sedation, motor dysfunction, hallucination, and dysphoria.145 G protein-biased agonists of κ-OR,146 including RB-64,145 mesyl salvinorin B, triazole 1.1, diphenethylamines and LOR17,141 were reported to minimize the adverse effects in preclinical settings. One of such, nalfurafine, was approved in Japan (2015) as an anti-pruritic agent for patients with chronic liver diseases.147
Carvedilol, known as a β1 and β2 adrenoceptor blocker, was found to be biased toward β-arrestin recruitment, G protein-coupled receptor kinase activation, and ERK1/2 phosphorylation. Joining its rank included alprenolol, bucindolol, and nebivolol, all are used to treat hypertension and congestive heart failure.148 In the case of β3 adrenoceptor, CL316243 is cAMP-biased, whereas L748337 and SR59230 are ERK/p38 phosphorylation-biased.149,150 Interestingly, CL316243 was also tested for treatment of obese mice.151,152 However, none of them have advanced to the clinic.
In contrast to µ-OR, arrestin bias is desirable for AT1R to improve cardiac performance.153 Nonetheless, clinical development of AT1R modulators either resulted in a phase IIb trial failure (TRV027) in 2017154 or never reached to clinical stage (SBpa, SVdF, SI, sarmesin, saralasin, and SII).155 Of note is that biased molecules may show species preference. For instance, CL316243 is more active in mice than in humans,156 whereas nalfurafine works better in humans vs. rodents.157 A list of therapeutic agents with biased signaling approved or advanced to clinical trials is shown in Table 5.
Allosteric modulation
In recent years, studies on allosteric GPCR modulators have gained unprecedented momentum.158,159,160,161 An allosteric modulator is a ligand binding to a position other than the orthosteric site but can modify responses of a receptor to stimulus. Allosteric modulators that enhance agonist-mediated response are called PAMs, while those attenuate the response are called NAMs. This phenomenon is very common such that the Allosteric Database 2019 (ASD, http://mdl.shsmu.edu.cn/ASD)162 records 37520 allosteric modulations on 118 GPCR members, covering all four classes.
Allosteric modulation is advantageous in terms of (i) using highly druggable pockets. In some cases, it is easier to design ligands at an allosteric site than the orthosteric site, such as class B GPCRs with orthosteric pockets wide open. For example, both PAM163 and NAMs107 binding to the same position at the TMD of GLP-1R were reported; (ii) improving selectivity. The orthosteric site and cognate ligand are often highly conserved, making it hard to discover very selective orthosteric binders. Meanwhile, non-conserved allosteric sites would be a better choice evidenced by discovery of many subtype selective allosteric modulators of acetylcholine102,164 and cannabinoid receptors165,166; (iii) introducing signal bias. Allosteric modulators with biased signaling were developed for prostaglandin F2α receptor167 and chemokine receptor CXCR4.168 Albeit still as an emerging concept, allosteric modulators have exhibited a great potential with some compounds being marketed or in clinical trials.160
However, developing allosteric modulators of GPCRs remains challenging—molecules recorded in the ASD largely concentrate on two subfamilies, the mGluRs (8 members, 17,115 modulations), and mAChRs (5 members, 7666 modulations), accounting for nearly 2/3 of the total number. Some individual receptors also contribute a significant proportion, such as CB1 (1948 modulations), GABAB (1286 modulations), and follicle-stimulating hormone receptor (1233 modulations). Excluding these “easy cases,” allosteric modulators are few in number. Furthermore, the structural diversity of the allosteric modulators is quite low, for many derivatives would be included soon after a parent compound is identified. The difficulty in developing allosteric modulators is partly due to the limitation of detecting allosteric behavior: Not every newly discovered active compound could be tested for its effect on binding affinity or EC50 of an orthosteric agonist, therefore some allosteric modulators were not correctly identified. For instance, BPTU in P2RY1, the first GPCR NAM solved in complex structure (PDB code: 4XNV),169 was not considered allosteric until the structure was obtained. To make things worse, NAMs may weaken the binding of an endogenous ligand thus behaving like a competitor, such as NDT9513727 in C5AR1170 (PDB code: 5O9H).171
The most effective way to identify the binding site of an allosteric modulator on a GPCR is solving the complex structure. Crystallography is an effective technique, while rapidly deployment of cryo-EM has started to deliver its promise (PDB codes: 6OIK172 and 6U1N173). To date, 17 GPCRs have reported structures in complex with allosteric modulators. Detailed analysis of complex structures before October 2018 was reported previously,161 and here we focus on insights provided by newly published results. The most unusual allosteric-binding sites on GPCRs are at the lipidic interface embedded in cell membrane. Five different positions were identified by crystal structures (Fig. 9): UP12, UP34, LOW34, LOW345, and LOW67. Four of them were recently reviewed.161 The LOW34 site was reported in 2019 for ORG27569 in CB1 (PDB code: 6KQI166; Fig. 10a).

Schematic diagram of allosteric sites at the lipidic surface identified by complex structures. The binding sites are manually labeled on the crystal structure of β2AR (PDB code: 6OBA180). Solid line, allosteric site at front side; dashed line, allosteric site at back side. UP, upper part aka close to the extracellular end; LOW, lower part aka close to the cytoplasmic end; numbers, main interacting transmembrane helices

Binding sites of allosteric modulators in GPCRs reported after October 2018, in comparison with related ligands. a NAM ORG27569 in CB1 (PDB code: 6KQI166) in comparison with cholesterol (PDB code: 5XRA179); b NAM AS408 (PDB code: 6OBA180) and PAM Cmpd-6FA (PDB code: 6N48181) in β2AR, in comparison with NDT9513727 in C5AR1 (PDB code: 6C1Q182) and PAM AP8 (PDB code: 5TZY183); c NAM maraviroc in CCR5 (PDB code: 4MBS187) in comparison with chemokine analog antagonist [5P7]CCL5 (PDB code: 5UIW188) and HIV envelope glycoprotein gp120 (PDB code: 6MEO189); d PAM TT-OAD2 in GLP-1R (PDB code: 6ORV190) in comparison with GLP-1 (PDB code: 5VAI191)
ORG27569 attracted much attention for its distinctive function: increasing the binding of orthosteric agonist CP55940 but making it act as inverse agonist.165 Many attempts were made to locate the binding site of ORG27569 by mutagenesis but the results are conflicting: one study showed that the effect of ORG27569 on CP55940-induced [35S]GTPγS binding was disturbed by mutations to multiple residues at the orthosteric site, leading to a hypothesis that ORG27569 stays in the same pocket close to CP55940.174 Another study found that ORG27569 reduced the binding of a fluorescence-labeled orthosteric antagonist, and the effect was only disturbed by mutations at the lipidic interface close to the cytoplasmic end of CB1.175 Besides, it was reported that the functions of ORG27569 were also affected by breaking a disulfide bond at the N-terminus176 or by constitutive active/inactive mutations at the cytoplasmic interface.174,175,177,178 The crystal structure exhibited that the position of ORG27569 is considerably overlapped with a cholesterol captured in another intermediate state (PDB code: 5XRA,179 Fig. 10a), consistent with the site located by the fluorescence-labeled orthosteric antagonist.175 At this site, the higher selectivity to CB1 over CB2 could be explained. Interestingly, ORG27569 is the only allosteric modulator at lipidic interface forming no hydrogen bond to the receptor.
There have been three more complex structures of allosteric modulators at lipidic interface since October 2018, all obtained by crystallography. Two are β2AR, with a NAM AS408 (PDB code: 6OBA180) or a PAM Cmpd-6FA (PDB code: 6N48181). Both allosteric modulators bind to the LOW345 site (Fig. 10b). The NAM stays at a position very similar to NAMs in C5AR1 (PDB codes: 5O9H,171 6C1R, and 6C1Q182) but the PAM is close to ICL2 and only partially overlaps with PAMs of FFAR1 (PDB codes: 5TZY183 and 5KW2184), showing a complex regulation nature at this site. The other complex structure is full-length GLP-1R with PF-06372222 (PDB code: 6LN2185), a NAM previously used to co-crystallized with GLP-1R TMD (PDB code: 5VEW107).
Even around the position of orthosteric ligands (among the helices and facing extracellular side), another ligand may occupy the space not taken by the endogenous ligand and act as an allosteric modulator. The very abundant PAMs/NAMs of mAChRs function in this mechanism. PAM LY2119620 in M2R (with the orthosteric agonist iperoxo and stabilized by a nanobody) was the first allosteric modulator to obtain complex structure with a class A GPCR (PDB code: 6MQT102). Recently, LY2119620 was also observed in protein complexes of M2R with G protein (PDB code: 6OIK172) or arrestin (PDB code: 6U1N173) by cryo-EM.
CCR5 is a chemokine receptor and an important anti-HIV drug target. A marketed inhibitor, maraviroc, has long been recognized as a NAM of CCR5. There were hypotheses that small molecule NAMs, chemokine, and the HIV-binding protein have separate binding sites.186 However, structures of CCR5 in complex with maraviroc (PDB code: 4MBS187), chemokine analog antagonist (PDB code: 5UIW188), or HIV envelope glycoprotein (PDB code: 6MEO189) show that these ligands highly overlap in CCR5 pocket (Fig. 10c). Therefore, the noncompetitive behavior of maraviroc may be due to a very extensive interface of peptidic CCR5 agonist, thus a small molecule cannot diminish the binding even with this much collision. The results illustrate that allosteric behavior is not equal to totally separated binding positions, because partially overlapped sites with different key interactions are also allowed.
The last case of allosteric modulator in extracellular pocket is PAM TT-OAD2 of GLP-1R (PDB code: 6ORV190). This small molecule agonist only slightly collides with the endogenous peptide (PDB code: 5VAI191, Fig. 10d), consistent with its behavior that only partially displaces an orthosteric probe.190
The cytoplasmic interface, where a GPCR interacts with intracellular partners, including Gα and β-arrestin, contains pockets suitable for drug design. So far, four small molecules have been validated by crystallography to bind at this position. The targets are three chemokine receptors (CCR2, PDB code: 5T1A192; CCR7, PDB code: 6QZH193; and CCR9, PDB code: 5LWE194) and β2AR (PDB code: 5X7D195). These ligands are all NAMs and proximately share the same binding site (TM1, TM2, TM6, TM7, ICL1, and H8). Their binding position does not overlap with Gα, therefore they may stabilize the inactive state by blocking conformational changes required for receptor activation. This site is generally non-conserved in the GPCR superfamily, thus targeting here may provide some selectivity. Additionally, many nanobodies at the cytoplasmic interface were also developed for several receptors, including AGTR1 (PDB codes: 6DO1196 and 6OS0197), β1AR (PDB code: 6IBL198), β2AR (PDB code: 6N48181), and SMO (PDB code: 6O3C114; for information before October 2018, see review161).
Multi-domain regulation is an interesting topic in allosteric modulator discovery. Class C GPCRs use ECDs to recognize their cognate ligands, leaving the classic pocket of TMD for allosteric modulating.161,199 This is the major reason why this class has a large number of allosteric modulators. In the case of mGluRs, both PAMs and NAMs have been widely reported, but only NAMs obtained complex structures—there is no solved active state structure. The full-length structures of mGlu5 (PDB codes: 6N51 and 6N5251) displayed how the binding of orthosteric agonist to ECD triggers the change of interaction between two monomers, but the conformational change of TMD remains elusive.
SMO in class F is also a multi-domain receptor. The first reported ligand of SMO cyclopamine (an antagonist causing birth defects) binds to the classic TMD pocket (PDB code: 4O9R200) shared by several other antagonists with different chemical scaffolds and an agonist (SAG).62,113,201,202 ALLO-1, an antagonist identified as allosteric modulator not competitive to cyclopamine, was recently found to bind at a deeper position in the pocket by photo-affinity labeling combined with mass spectrometry (MS).203 SMO has another pocket in ECD that interacts with steroids, including cholesterol (PDB codes: 5L7D113 and 6D35204) and 20(S)-hydroxycholesterol (PDB code: 5KZV119). Since cholesterol has been the most favored candidate of SMO endogenous ligand, the ECD pocket is treated as orthosteric making the TMD pocket allosteric. However, newly obtained structures demonstrated that cholesterol or its analog can also bind to TMD pocket (PDB codes: 6O3C114 and 6OT0115), leaving the question open for which is the true orthosteric site.
Disease indication
GPCRs are involved in many human diseases and specific drug intervention is one of the most celebrating achievements in the pharmaceutical industry (Table S3 and Fig. S1). Among all available drugs targeting GPCRs, HRH1, DRD2, M1R, and ADRA1A are the most frequently addressed for indications such as hypertension, allergy, pain, and schizophrenia, and 33% of them have >1 indication with an overall average of 1.5. Although CNS diseases are still popular accounting for 26% of all approved indications, development focuses have now been shifted to T2DM, obesity, multiple sclerosis, smoking cessation, short bowel syndrome, and hypocalcemia. Repurposing of existing drugs for new indications also emerged to supplement discovery efforts.
Structure-based drug design
As two general types of computer-aided drug design techniques (Table 6),205 SBDD and ligand-based drug design, exploit the structural information of protein targets and the knowledge of known ligands, respectively (Table 6). SBDD, on the basis of crystal/cryo-EM/NMR structures or homology models, first identifies key sites and important interactions responsible for target functions, then screens large virtual library/designed agents that disrupt or enhance such interactions to modulate relevant biological processes and/or signaling pathways by molecular docking, and finally discovers active leads with desired pharmacological properties.
Clearly, the past decade is a golden age for SBDD on GPCR. With the year of 2011 (when LPC crystallization,206 fusion proteins,8 and other key techniques collaborated to launch the outbreak of GPCR structure determination including the landmark β2AR-Gs) for watershed,207 SBDD of GPCR evolves two distinct stages: rhodopsin-based homology model and truly authentic structure of individual receptors. Boosted by the fast-increasing number of high-quality GPCR structures, improved accuracy of combinational computational approaches, and better understanding of activation mechanism and pharmacology, SBDD is developing rapidly with fruitful scientific reports and increasing GPCR-targeted drugs contributed by this approach. Considering the length of time required for a drug to be available on the market (10–15 years) and the chance of applying structural biology information to hit discovery and lead optimization in the first 2–3 years of a drug discovery program, it is probably too early to see the approval of GPCR-targeted drugs being developed with the aid of a structure, and such situation is likely to change as the tremendous efforts from both academia and industry start to bear the fruits of successes. The following is a brief account of recent advances in three main aspects of SBDD (chemical space, receptor dynamics, and pose evaluation) in the context of their application in GPCR pharmacology.
Optimized virtual library
Despite the vast chemical space (>1063 drug-like molecules), only a nominal fraction has been explored by SBDD, where both the compound availability and insufficient diversity limited the number of screened ligands. To overcome these problems, ultra-large208,209,210 and focused libraries211,212,213 were employed. Lyu and colleagues208 presented an excellent model of “bigger is better” in virtual drug screening. Based on the 130 well-characterized reactions, they generated 170 million make-on-demand compounds (http://zinc15.docking.org/), the resulting library is remarkably diverse with >10.7 million scaffolds unavailable before. By docking 138 million molecules against DRD4, they discovered 81 new chemotypes (24% hit rate), 30 of them showed submicromolar activity, including a 180-pM subtype-selective and Gi-biased DRD4 agonist. This ultra-large library docking study provides important information: (i) hit rate fell almost monotonically with docking score; (ii) hit rate vs. score curve of DRD4 predicted that 1 from every 873 compounds may have a minimum affinity of 1 μM; and (iii) human visual evaluation improved the selected compound with higher affinities, efficacies, and potencies but not the hit rate. A follow-up study on MT1 by docking >150 million “lead-like” molecules209 identified 15 active leads (39% hit rate) with potencies ranging from 470 pM to 6 μM. Alternatively, focused library210,211,212,213,214,215 including scaffold library, natural products, dark chemical matter (i.e., chemicals that have never shown bioactivity tested in over 100 assays), and fragment- and lead-like libraries were introduced in virtual screening (VS) for dozens of receptors. Focused on compound library of traditional Chinese medicine (TCM), Liu et al. found that salvianolic acids A and C antagonized the activity of both P2RY1 and P2RY12 purinoceptors in the low µM range, while salvianolic acid B antagonized the P2RY12 purinoceptor. Remarkably, these three salvianolic acids are major active components of the broadly used hemorheologic TCM Danshen (Salvia militorrhiza). Taking NTSR1 as an example, Ranganathan et al. found that the fragment library tended to have higher hit rate than that of the lead-like library (19%) but the affinities were ∼100-fold weaker. Collectively, these results demonstrate the importance and advantages of ultra-large and tailored libraries in discovering potent GPCR modulators.
Receptor dynamics
Emerging evidence from crystallography, spectroscopy, and molecular dynamics (MD) simulations have demonstrated the crucial roles of GPCR dynamics involved in ligand recognition, receptor activation, and allosteric modulation.216,217,218 To consider the protein flexibility during GPCR-related SBDD, many computational approaches217 including rotamer sampling, induced-fit docking, and ensemble docking have been employed showing a great promise, especially in the search of biased, bi-topic, or allosteric modulators. During ensemble docking,212,217,219,220,221 ligands are docked into multiple structures representing different possible conformational states rather than a single structure, where the targets could be multiple crystal structures or extracted from MD/Monte Carlo (MC) simulations or normal mode analysis (NMA). By evaluating the known ligand enrichment, as well as selectivity for agonists or antagonists on seven GPCR/ligand co-structures, Coudrat et al. found that small variations in structural features are responsible for their success in VS, while a combination of ligand/receptor interaction patterns and predicted interaction strength is associated with the predictive power of binding pockets in VS.220 Compared to the Glide VS workflow, the combination of accelerated MD simulations and Glide induced fit docking of M2R by Miao et al. provided much-improved enrichment factors and identified four PAMs and one NAM with unprecedented chemical diversity.221 For 5-HT1A whose crystal structure is not available, Warszycki et al. applied MC and NMA to generate an ensemble of binding pockets with the input of a homology template and known active compounds and finally discovered two new active ligands through VS.222
Pose evaluation
Correctly selecting and ranking poses of docked compounds in the ligand-binding pockets have been a challenge for SBDD, especially for GPCR that is embedded in the cell membrane with significant conformational adaptability. To address this problem, many physics-based scoring functions223,224,225,226 integrated with some user-friendly computer programs (e.g., Dock, GOLD, AutoDock, Glide, and rDock) were routinely adopted in SBDD. Recently, precise computational approaches including free energy calculation methods like molecular mechanics/Poisson–Boltzmann surface area (MM/PB(GB)SA),227,228 free energy perturbation (FEP),229 quantum mechanical/MM calculations,230 and fragment molecular orbital231,232 have been employed with improved performance. Compared to the empirical scoring functions, MM/PB(GB)SA and FEP are physically more rigorous free-energy calculation methods with an increased computational cost and have been adopted in the studies of DNA–ligand, protein–ligand, and protein–protein interactions.233,234 By introducing of the minimization-based MM/GBSA refining and rescoring of docked poses, Zhou et al. identified seven 5-HT2B antagonists with novel chemical scaffolds and the most potent one has an IC50 of 27.3 nM in a cellular assay.227 Lenselink et al. used FEP to design A2AR antagonists and identified a highly potent molecule with Ki of 1.2 nM.232 However, computational investigation across 20 class A crystal structures and 934 known ligands demonstrated that the correlations between predicted binding free energy by MM/PBSA and experimental data varied significantly. The observed variations exist between individual receptors and are highly system specific,235 indicating that successful application of MM/PBSA may require additional efforts in validation of experimental data and optimization of simulation/calculation parameters. Alternatively, protein–ligand interaction fingerprints exacted from available crystal structures225,236 fueled docking score with protein–ligand-binding mode information and resulted in improved VS virtual hit rates for β2AR (53%) and HRH1 (73%) with up to nM affinities and potencies.225,236
Collectively, innovation in VS and pose evaluation, along with evolution of computational hardware, has significantly advanced SBDD and is expected to lift the discovery efficiency to a new height, since GPCRs have multiple downstream signaling pathways responsible for distinct functions or consequences. The high degree of sequence and pocket similarities between different subtypes demands for novel ligands with superior specificity and selectivity. In this regard, allosteric and biased modulators may offer additional pharmacological benefits.
Subtype selectivity
It is known that GPCR subtypes share high sequence identities in orthosteric sites with distinct distribution and downstream signaling profiles. Cross-reactivity among subtypes could cause undesired side effects. For example, five MR subtypes display different G protein coupling features (Gq/11: M1R, M3R and M5R; Gi/o: M2R and M4R) and organ distribution (CNS, M1R; peripheral tissues such as heart and colon, M2R), while their sequence identify (64–82%) and similarity (82–92%) in TMD are quite conserved.172 Similar observations were seen among dopamine receptors (DRD1 to DRD5), histamine receptors (HRH1 to HRH4), and adenosine receptors (A1AR, A2AR, A2BR, and A3AR). Guided by structural information, rational design of subtype selective compounds progresses steadily. Using an extended DRD2-specific binding pocket from the haloperidol-bound DRD2 crystal structure, Fan et al. discovered two highly selective DRD2 agonists (O4SE6 and O8LE6) that specifically activate DRD2 (EC50 = 1 µM) after screening of 320 non-olfactory GPCRs.237 Through VS of 3.1 million molecules against M2 and M3, Kruse et al. identified a partial M3 agonist without measurable M2 agonism, capable of stimulating insulin release from a mouse β-cell line.238 Wei et al. reported a multistage VS of the ChemDiv library (1,492,362 compounds) toward A1AR and discovered four novel antagonists with good affinity and selectively (>100-fold) over A2AR.239
Biased signaling
Recently, Suomivuori et al. performed extensive MD simulations to identify two major signaling conformations that couple effectively to arrestin or G protein, respectively. They then designed ligands via minor chemical modification resulting in strong arrestin-biased or Gq-biased signal transduction.240 Meanwhile, McCorvy and colleagues discovered that specific ECL2–ligand contacts are associated with β-arrestin recruitment, whereas blockage of TM5 interaction reduces the Gi/o signaling. An arrestin-biased DRD2 modulator was thus made exhibiting a calculated bias factor of 20 relative to quinpirole.241 In addition, Mannel et al. conducted a VS of a tailored virtual library bearing 2,3-dichlorophenylpiperazine for DRD2 and found that 18 compounds occupy both orthosteric and allosteric sites, and 4 of them stimulated β-arrestin recruitment (EC50 = 320 nM, Emax = 16%) without detectable G protein signaling.214
Allosterism
In the past 5 years, an increasing number of receptor–allosteric modulator complex structures revealed diversified positions of allosteric sites and a variety of binding modes, thereby deepening our understanding of allosteric modulation in terms of underlying mechanisms and structural basis. Further to conventional orthosteric ligands, allosteric modulators affect receptor function in different ways. While PAM may enhance maximal efficacy, NAM could reduce agonist signaling strength.159,160,242 It was reported that a PAM of M2R is located above the orthosteric site and interacts with ECLs. Korczynska et al. screened 4.6 million molecules against the allosteric sites of M2R and identified a PAM that potentiated the action of antagonist N-methyl scopolamine (NMS). Subsequent optimization led to a subtype-selective compound 628 that increased NMS binding with a co-operativity factor of 5.5 and a KB of 1.1 µM.16 Alternatively, Lückmann et al. carried out MD simulations of agonist-removed FFAR1 and found that closure of a potential allosteric site is associated with agonist binding—compounds that bind to this site to prevent the closure functions as allosteric agonists.243 Obviously, aided by >400 structures from 82 receptors, SBDD is now entering into a new era with substantial knowledge of GPCR signaling103,110,244 and drug candidate attributes.245
Novel screening technology
As GPCRs represent the most prominent family of therapeutic targets,132 innumerable efforts have been made in both industry and academia to screen for novel ligands that can modulate the activity of a specified GPCR and serve as lead compounds for drug development.
A diverse array of experimental technologies suitable for assaying protein–ligand interactions have been directly applied or tailored to GPCR-targeted ligand screening, and they can be classified into three main categories: binding-based, stability-based, and cell signaling-based assays (Table 7). Binding-based assays monitor the physical interactions between a GPCR protein typically in a purified recombinant form with individual test compounds. Cell signaling-based assays measure downstream effectors (e.g., cAMP, Ca2+, IP1/IP3) of specific intracellular signaling pathways known to be mediated by GPCR, which reflect the functional outcome of ligand binding to the receptor. Stability-based assays assess the variation of thermal stability for a purified protein when treated by test compounds. These different techniques vary in the ligand screening throughput and binding characteristics (Table 7). In the lead discovery stage, both binding- and signaling/activity-based assays are implemented in a parallel or sequential manner, as the multipronged use of complementary techniques would reduce the overall false-positive and false-negative rates.246
Here we summarize about 20 experimental screening technologies adapted to GPCR ligand discovery (Table 7) and highlight the most recent development of binding-based approaches. Notably, an update of assays assessing GPCR activation and signaling has been provided in a previous review247 and will not be elaborated here. The structure-based VS is covered in the above section. Structural elucidation technologies (e.g., X-ray crystallography, single-particle cryo-EM, NMR, and HDX-MS) not suitable for high-throughput screening (HTS) are also excluded.
DNA-encoded library (DEL)
Impressive technological advances have been made for binding-based ligand screening over the past decade. Specifically, DEL has emerged as a powerful approach to drug discovery.248,249,250,251 Created by split and pool synthesis, DEL usually contains hundreds of thousands to billions of distinct small molecule–DNA conjugates. A majority of DEL-based HTS reported to date involve incubation of an immobilized target protein with the library before the protein–ligand complexes are isolated. Encoding DNA tags associated with the immobilized target are then amplified and sequenced to assign relevant chemical structures.250,251 Although DEL was predominantly applied to ligand screening against soluble proteins such as enzymes, successful adaptation of this technique to GPCRs was reported in a few cases.252,253,254,255 Lefkowitz group reported the discovery of a NAM for β2AR by screening a DEL of 190 million.252 This NAM not only has a unique chemotype but also exhibits low μM affinity and inhibits cAMP production as well as β-arrestin recruitment. Later on, the same team discovered the first small molecule PAM for β2AR through HTS of >500 million DEL compounds.253 Both NAM and PAM demonstrated high selectivity. Of note is that the NAM was found using unliganded β2AR, whereas the PAM was unmasked via intentional application of β2AR with its orthosteric site occupied by an agonist thereby shifting the receptor to the active state.252,253 These two studies elegantly demonstrated a proof-of-concept strategy for binding-based screening of allosteric modulators targeting different conformational states.253
The power of DEL in the discovery of allosteric GPCR modulators was further demonstrated for PAR2.254 Screening a billion-size library with a thermostabilized PAR2 mutant resulted in the identification of several agonists and antagonists, and some of them bind to an allosteric pocket in the TMD of PAR2. A similar approach was used to discover tachykinin receptor neurokinin-3 (NK3) antagonists of low nM potency involving NK3 overexpressing cells and a library containing tens of millions DNA-encoded compounds.255 Clearly, DEL-based ligand screening against GPCRs and other integral membrane proteins offers great promises as it circumvents difficulties in receptor purification.
Affinity selection MS
Due to the high sensitivity and high selectivity of modern MS for both protein and small molecule analysis, versatile MS-based technologies have been developed in the past two decades for screening ligands of a given protein target or characterization of ligand-binding properties (Table 7). Almost all of them were originally developed for measuring ligand interactions with soluble proteins,256,257,258,259,260,261 and recently they have been adapted to more challenging GPCR drug discovery. The majority of MS-based technologies (e.g., automated ligand identification system (ALIS), ultrafiltration–liquid chromatography/MS, frontal affinity chromatography–MS, membrane-based affinity MS, and competitive MS binding) employ a methodology very similar to DEL as they all capture and detect ligands physically associated with a given GPCR,262,263,264,265,266,267 except that native MS analyzes the entire ligand-bound receptor complexes.268,269 In general, these methods have several advantages over ligand binding or cell signaling assays: (i) unbiased and direct detection of ligand–receptor binding facilitates the identification of both orthosteric and allosteric modulators; (ii) confirmation of ligand identity with accurate mass measurement; (iii) no chemical labeling or DNA encoding of test compounds; and (iv) quantitative MS analysis enables ranking of ligand affinity or evaluation of binding characteristics.
ALIS is currently the most prevailing MS-based technique employed in pharmaceutical companies for HTS of large-scale synthetic compound libraries.256,261,270,271,272,273 This system integrates size exclusion chromatography for isolating protein–ligand complexes and reverse-phase chromatography for dissociating bound ligands, which are then identified by high-resolution MS. Not surprisingly, the application of ALIS to ligand screening for GPCRs substantially lagged behind soluble proteins due to difficulties in obtaining membrane receptors of sufficient purity and stability. The earliest application was ligand screening for M2R,274 in which purified M2R was incubated with a 1500-compound pool in each round of affinity selection. After screening a total of 350,000 compounds, one orthosteric antagonist and one allosteric modulator were identified for AChR. Later on, a similar strategy was implemented to screen ligands for CXCR4 using two libraries comprised of 48,000 and 2.75 million compounds, respectively. Each reaction consumed 250 ng purified receptor incubated with a pool of 100 or 2000 compounds. Out of the 362 primary hits, 34 were subsequently confirmed to be new antagonists.262
Membrane-based affinity MS developed by Shui’s group enables ligand screening toward wild-type active GPCRs embedded in the cell membrane.266 It features isolation of membrane fractions from cells expressing a GPCR at high yield and incubation of the cell membrane with a compound cocktail, thus keeping the receptor in its native conformation and eliminating the need of protein purification. Compounds associated with the receptor were then released and subjected to high-resolution MS for structural assignment (Fig. 11a). Each incubation consumed about 2 µg membrane-embedded GPCR protein with a pool of 480 compounds. Primary hits were selected based on the binding index (BI) derived from quantitative MS signals used to distinguish putative ligands from non-specific binders (Fig. 11b). Screening a small compound library with this approach led to the discovery of an antagonist for the 5-HT2C receptor and four PAMs for GLP-1R that are not reported previously266 (Fig. 11c).

Developing affinity MS approaches for GPCR ligand screening. a Experimental workflow of membrane-based affinity MS. b Membrane-based affinity MS screening of 4333 compounds split into 9 cocktails against GLP-1R. Initial hits are indicated by red dots, while gray dots represent negatives. c Binding of one new ligand to 5-HT2C (upper) and four new ligands to GLP-1R (lower) were validated by a radioligand-binding assay. d Experimental workflow of affinity MS-based screening of natural herb extracts. e Initial hits from screening fractionated herbal extracts toward 5-HT2C. Aporphines are annotated with larger pink dots. BI binding index. f Structural validation of 1857 by MSMS analysis. g 1857 displayed selective agonism at 5-HT2C. Source: adapted from Qin et al.266 and Zhang et al.282
More recently, the same team devised another affinity MS strategy that enabled screening of 20,000 compounds in one pool.275 Specifically, they modified the workflow by performing iterative rounds of affinity selection for compounds associated with A2AR. Similar to the previously described single-round affinity MS screening assay, quantitative measurement of BI renders detection of high-affinity ligands in this experiment. By comparing the selection of 16 benchmark A2AR ligands from screening compound pools of 480-mix, 2400-mix, 4800-mix, and 20K-mix, they demonstrated that this accelerated affinity MS screening approach, using either the purified receptor or receptor-expressing cell membranes, allowed detection of most high-affinity A2AR ligands (Kd or Ki <5 µM), and significant reduction of protein consumption and MS instrument time.275 Three new antagonists for A2AR were identified as a result. It is likely that the throughput of this method could be further increased to assay close to or above 1 million compounds in one pool.275
The affinity MS technique has been widely employed to fish out and identify putative ligands toward various enzyme targets from complex extracts of natural products, which could promote lead discovery from TCM.276,277,278,279,280,281 Indeed, this technique was successfully extended to GPCR ligand screening from herbal extracts. It involved the optimization of receptor construct and integration of affinity MS with metabolomics data mining workflow for sensitive and accurate ligand identification282 (Fig. 11d). After screening a panel of herbal extracts, a naturally occurring aporphine compound (1857) displaying strong subtype selectivity for 5-HT2C without affecting 5-HT2A or 5-HT2B was discovered (Fig. 11e–g). Moreover, this new lead exhibited exclusive bias toward G protein signaling and showed in vivo efficacy for food intake suppression and weight loss.282
Although not directly applied to GPCRs, a previously reported cell-based assay vascular endothelial growth factor receptor 2 (VEGFR2) is interesting.283 It used a special one-bead-one-compound library of peptoids and cells overexpressing VEGFR2. Beads bound to the color-coded VEGFR2-expressing cells were selected under fluorescence microscopy and the attached ligands decoded by tandem MS analysis. Hits with low μM affinity to the soluble VEGFR2 ectodomain were identified subsequently. We envision that these membrane-based or cell-based screening platforms will make a major impact on GPCR drug discovery, especially when they are fully integrated.
Competitive MS binding assay employs a non-radioactive ligand to compete the binding of a test compound to a protein target. It resembles radioligand-binding assays but avoids the use of radioisotope.284,285,286 When assaying, the marker ligand liberated from the target is measured by an multiple reaction monitoring-based MS method of high sensitivity and selectivity for compound detection. Not only established for a number of transporters and ion channels,287,288,289 this approach is equally effective in addressing GPCRs as recently exemplified on A1AR/A2AR and DRD1/2/5.267,285,290,291 It was shown that unlabeled marker compounds could substitute their radiolabeled counterparts in all types of ligand-binding characterization studies, including saturation, displacement, dissociation, and competitive association, yielding results in excellent accordance with classic radioligand-binding assays.267,290
Emerging opportunities and prospects
Recent scientific and technological advancements in GPCR biology have provided an enormous amount of information that will benefit our current and future efforts in rational drug design. Integration and refinement of massive data by artificial intelligence is a clear direction to guide both virtual and experimental screening of efficacious therapeutic agents with new scaffolds and of novel chemotypes for all classes of GPCRs.
However, as described in this review, factors that influence GPCR drug discovery include, but not limited to, therapeutic target, chemical diversity, mechanism of signaling, ligand-binding site, mode of action, clinical indication, polypharmacology, etc. Future opportunities may arise from: (i) de-orphanization of orphan GPCRs to provide novel targets; (ii) new indication for drug intervention via discovery and/or repurposing efforts; (iii) development of lead compounds targeting classes B2 and F GPCRs to address unmet medical needs; and (iv) validation of polypharmacology may lead to improved drug therapies.
References
Insel, P. A. et al. GPCRomics: an approach to discover GPCR drug targets. Trends Pharmacol. Sci. 40, 378–387 (2019).
Sriram, K. & Insel, P. A. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 (2018).
Wootten, D. et al. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19, 638–653 (2018).
Hauser, A. S. et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Shimada, I. et al. GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 18, 59–82 (2019).
Dalesio, N. M., Barreto Ortiz, S. F., Pluznick, J. L. & Berkowitz, D. E. Olfactory, taste, and photo sensory receptors in non-sensory organs: it just makes sense. Front. Physiol. 9, 1673 (2018).
Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007).
Rosenbaum, D. M. et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273 (2007).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017).
Safdari, H. A., Pandey, S., Shukla, A. K. & Dutta, S. Illuminating GPCR signaling by cryo-EM. Trends Cell Biol. 28, 591–594 (2018).
Congreve, M., de Graaf, C., Swain, N. A. & Tate, C. G. Impact of GPCR structures on drug discovery. Cell 181, 81–91 (2020).
Wootten, D. et al. Allostery and biased agonism at class B G protein-coupled receptors. Chem. Rev. 117, 111–138 (2017).
Inoue, A. et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947. e1925 (2019).
Lane, J. R. et al. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 13, 929–937 (2017).
Manglik, A. et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016).
Korczynska, M. et al. Structure-based discovery of selective positive allosteric modulators of antagonists for the M2 muscarinic acetylcholine receptor. Proc. Natl Acad. Sci. USA 115, E2419–E2428 (2018).
Foster, S. R. et al. Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell 179, 895–908. e821 (2019).
Hu, G. M., Mai, T. L. & Chen, C. M. Visualizing the GPCR network: classification and evolution. Sci. Rep. 7, 15495 (2017).
Basith, S. et al. Exploring G protein-coupled receptors (GPCRs) ligand space via cheminformatics approaches: impact on rational drug design. Front. Pharmacol. 9, 128 (2018).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Alexander, S. P. H. et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 176, S21–S141 (2019).
Bhudia, N. et al. G protein-coupling of adhesion GPCRs ADGRE2/EMR2 and ADGRE5/CD97, and activation of G protein signalling by an anti-EMR2 antibody. Sci. Rep. 10, 1004 (2020).
Pal, K., Melcher, K. & Xu, H. E. Structure and mechanism for recognition of peptide hormones by class B G-protein-coupled receptors. Acta Pharmacol. Sin. 33, 300–311 (2012).
Bondarev, A. D. et al. Opportunities and challenges for drug discovery in modulating adhesion G protein-coupled receptor (GPCR) functions. Expert Opin. Drug Discov. 15, 1291–1307 (2020).
Vizurraga, A. et al. Mechanisms of adhesion G protein-coupled receptor activation. J. Biol. Chem. 295, 14065–14083 (2020).
Muller, T. D. et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130 (2019).
Sekar, R., Singh, K., Arokiaraj, A. W. & Chow, B. K. Pharmacological actions of glucagon-like peptide-1, gastric inhibitory polypeptide, and glucagon. Int. Rev. Cell Mol. Biol. 326, 279–341 (2016).
Yu, M. et al. Battle of GLP-1 delivery technologies. Adv. Drug Deliv. Rev. 130, 113–130 (2018).
Drucker, D. J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 27, 740–756 (2018).
Pratley, R. et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet 394, 39–50 (2019).
Blundell, J. et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab. 19, 1242–1251 (2017).
Williams, D. M., Nawaz, A. & Evans, M. Drug therapy in obesity: a review of current and emerging treatments. Diabetes Ther. 11, 1199–1216 (2020).
Knerr, P. J. et al. Selection and progression of unimolecular agonists at the GIP, GLP-1, and glucagon receptors as drug candidates. Peptides 125, 170225 (2020).
Frias, J. P. et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 26, 343–352 e342 (2017).
Parker, V. E. R. et al. Efficacy, safety, and mechanistic insights of cotadutide, a dual receptor glucagon-like peptide-1 and glucagon agonist. J. Clin. Endocrinol. Metab. 105, dgz047 (2020).
Visentin, R. et al. Dual glucagon-like peptide-1 receptor/glucagon receptor agonist SAR425899 improves beta-cell function in type 2 diabetes. Diabetes Obes. Metab. 22, 640–647 (2020).
Tillner, J. et al. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes. Metab. 21, 120–128 (2019).
Armstrong, D. et al. Colon polyps in patients with short bowel syndrome before and after teduglutide: post hoc analysis of the STEPS study series. Clin. Nutr. 39, 1774–1777 (2020).
Gingell, J. J., Hendrikse, E. R. & Hay, D. L. New insights into the regulation of CGRP-family receptors. Trends Pharmacol. Sci. 40, 71–83 (2019).
Hay, D. L., Garelja, M. L., Poyner, D. R. & Walker, C. S. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br. J. Pharmacol. 175, 3–17 (2018).
Davenport, A. P. et al. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 19, 389–413 (2020).
Dolgin, E. First GPCR-directed antibody passes approval milestone. Nat. Rev. Drug Discov. 17, 457–459 (2018).
Edvinsson, L., Haanes, K. A., Warfvinge, K. & Krause, D. N. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat. Rev. Neurol. 14, 338–350 (2018).
Ishida, J. et al. Growth hormone secretagogues: history, mechanism of action, and clinical development. JCSM Rapid Commun. 3, 25–37 (2020).
Karageorgiadis, A. S. et al. Ectopic adrenocorticotropic hormone and corticotropin-releasing hormone co-secreting tumors in children and adolescents causing cushing syndrome: a diagnostic dilemma and how to solve it. J. Clin. Endocrinol. Metab. 100, 141–148 (2015).
Leder, B. Z. et al. Effects of abaloparatide, a human parathyroid hormone-related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J. Clin. Endocrinol. Metab. 100, 697–706 (2015).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006).
Niswender, C. M. & Conn, P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322 (2010).
Pin, J. P. et al. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 272, 2947–2955 (2005).
Geng, Y. et al. Structural mechanism of ligand activation in human GABA(B) receptor. Nature 504, 254–259 (2013).
Koehl, A. et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019).
Mao, C. et al. Cryo-EM structures of inactive and active GABAB receptor. Cell Res. 30, 564–573 (2020).
Papasergi-Scott, M. M. et al. Structures of metabotropic GABAB receptor. Nature 584, 310–314 (2020).
Park, J. et al. Structure of human GABAB receptor in an inactive state. Nature 584, 304–309 (2020).
Shaye, H. et al. Structural basis of the activation of a metabotropic GABA receptor. Nature 584, 298–303 (2020).
De Witte, P., Littleton, J., Parot, P. & Koob, G. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs 19, 517–537 (2005).
Messa, P., Alfieri, C. & Brezzi, B. Cinacalcet: pharmacological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 4, 1551–1560 (2008).
Ruat, M., Hoch, L., Faure, H. & Rognan, D. Targeting of smoothened for therapeutic gain. Trends Pharmacol. Sci. 35, 237–246 (2014).
Schulte, G. & Wright, S. C. Frizzleds as GPCRs - more conventional than we thought! Trends Pharmacol. Sci. 39, 828–842 (2018).
Zhang, X., Dong, S. & Xu, F. Structural and druggability landscape of Frizzled G protein-coupled receptors. Trends Biochem. Sci. 43, 1033–1046 (2018).
Yang, S. et al. Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 560, 666–670 (2018).
Zhang, X. et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 8, 15383 (2017).
Tsutsumi, N. et al. Structure of human Frizzled5 by fiducial-assisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. Elife. 9, e58464 (2020).
Schulte, G. Frizzleds and WNT/beta-catenin signaling–The black box of ligand-receptor selectivity, complex stoichiometry and activation kinetics. Eur. J. Pharmacol. 763, 191–195 (2015).
Yang, X. et al. Development of covalent ligands for G protein-coupled receptors: a case for the human adenosine A3 receptor. J. Med. Chem. 62, 3539–3552 (2019).
Weichert, D. & Gmeiner, P. Covalent molecular probes for class A G protein-coupled receptors: advances and applications. ACS Chem. Biol. 10, 1376–1386 (2015).
Ricart-Ortega, M., Font, J. & Llebaria, A. GPCR photopharmacology. Mol. Cell. Endocrinol. 488, 36–51 (2019).
Hull, K., Morstein, J. & Trauner, D. In vivo photopharmacology. Chem. Rev. 118, 10710–10747 (2018).
Muratspahic, E., Freissmuth, M. & Gruber, C. W. Nature-derived peptides: a growing niche for GPCR ligand discovery. Trends Pharmacol. Sci. 40, 309–326 (2019).
Drucker, D. J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 19, 277–289 (2020).
Davenport, A. P. et al. International union of basic and clinical pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol. Rev. 65, 967–986 (2013).
Hutchings, C. J. A review of antibody-based therapeutics targeting G protein-coupled receptors: an update. Expert Opin. Biol. Ther. 20, 925–935 (2020).
Hutchings, C. J., Koglin, M., Olson, W. C. & Marshall, F. H. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat. Rev. Drug Discov. 16, 787–810 (2017).
Kahsai, A. W. et al. Conformationally selective RNA aptamers allosterically modulate the beta2-adrenoceptor. Nat. Chem. Biol. 12, 709–716 (2016).
Yoon, S. & Rossi, J. J. Aptamers: uptake mechanisms and intracellular applications. Adv. Drug Deliv. Rev. 134, 22–35 (2018).
Limbird, L. E., Meyts, P. D. & Lefkowitz, R. J. Beta-adrenergic receptors: evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 64, 1160–1168 (1975).
Li, J. et al. Nongenetic engineering strategies for regulating receptor oligomerization in living cells. Chem. Soc. Rev. 49, 1545–1568 (2020).
Roecker, A. J., Cox, C. D. & Coleman, P. J. Orexin receptor antagonists: new therapeutic agents for the treatment of insomnia. J. Med. Chem. 59, 504–530 (2016).
Coleman, P. J. et al. The discovery of suvorexant, the first orexin receptor drug for insomnia. Annu Rev. Pharmacol. Toxicol. 57, 509–533 (2017).
Scott, L. J. Lemborexant: first approval. Drugs 80, 425–432 (2020).
Yoshida, Y. et al. Design, synthesis, and structure-activity relationships of a series of novel N-aryl-2-phenylcyclopropanecarboxamide that are potent and orally active orexin receptor antagonists. Bioorg. Med. Chem. 22, 6071–6088 (2014).
Yoshida, Y. et al. Discovery of (1R,2S)-2-{[(2,4-Dimethylpyrimidin-5-yl)oxy]methy1}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide(E2006): a potent and efficacious oral orexin receptor antagonist. J. Med. Chem. 58, 4648–4664 (2015).
Bell, I. M. Calcitonin gene-related peptide receptor antagonists: new therapeutic agents for migraine. J. Med. Chem. 57, 7838–7858 (2014).
Williams, T. M. et al. Non-peptide calcitonin gene-related peptide receptor antagonists from a benzodiazepinone lead. Bioorg. Med. Chem. Lett. 16, 2595–2598 (2006).
Rudolf, K. et al. Development of human calcitonin gene-related peptide (CGRP) receptor antagonists. 1. Potent and selective small molecule CGRP antagonists.1-[N-2-[3,5-dibromo-N-[[4-(3,4-dihydro-2(1H)-oxoquinazolin-3-yl)-1-piperidinyl]carbonyl]-D-tyrosyl]-L-lysyl]-4-(4-pyridinyl)piperazine: the first CGRP antagonistfor clinical trials in acute migraine. J. Med. Chem. 48, 5921–5931 (2005).
Shaw, A. W. et al. Caprolactams as potent CGRP receptor antagonists for the treatment of migraine. Bioorg. Med. Chem. Lett. 17, 4795–4798 (2007).
Paone, D. V. et al. Potent, orally bioavailable calcitonin gene-related peptide receptor antagonists for the treatment of migraine: discovery of N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxamide (MK-0974). J. Med. Chem. 50, 5564–5567 (2007).
Luo, G. et al. Discovery of BMS-846372, a potent and orally active human CGRP receptor antagonist for the treatment of migraine. ACS Med. Chem. Lett. 3, 337–341 (2012).
Luo, G. et al. Discovery of (5S,6S,9R)-5-amino-6-(2,3-difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyri din-9-yl 4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxylate (BMS-927711): an oral calcitonin gene-related peptide (CGRP) antagonist in clinical trials for treating migraine. J. Med. Chem. 55, 10644–10651 (2012).
Palczewski, K. et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 (2000).
Hanson, M. A. et al. Profiling of membrane protein variants in a baculovirus system by coupling cell-surface detection with small-scale parallel expression. Protein Expr. Purif. 56, 85–92 (2007).
Chae, P. S. et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008 (2010).
Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180 (2011).
Rasmussen, S. G. et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 (2007).
Caffrey, M. Crystallizing membrane proteins for structure-function studies using lipidic mesophases. Biochem. Soc. Trans. 39, 725–732 (2011).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Kang, Y. et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015).
Katritch, V., Cherezov, V. & Stevens, R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013).
Kruse, A. C. et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556 (2012).
Zhang, H. et al. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem. 290, 29127–29139 (2015).
Kruse, A. C. et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013).
Zhou, Q. et al. Common activation mechanism of class A GPCRs. Elife. 8, e50279 (2019).
Karageorgos, V. et al. Current understanding of the structure and function of family B GPCRs to design novel drugs. Hormones 17, 45–59 (2018).
Hollenstein, K. et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 (2013).
Jazayeri, A. et al. Extra-helical binding site of a glucagon receptor antagonist. Nature 533, 274–277 (2016).
Song, G. et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315 (2017).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Dore, A. S. et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 (2014).
Thal, D. M., Glukhova, A., Sexton, P. M. & Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018).
Wu, H. et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 (2014).
Schulte, G. International Union of Basic and Clinical Pharmacology. LXXX. The class Frizzled receptors. Pharmacol. Rev. 62, 632–667 (2010).
Byrne, E. F. X. et al. Structural basis of smoothened regulation by its extracellular domains. Nature 535, 517–522 (2016).
Deshpande, I. et al. Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 571, 284–288 (2019).
Qi, X. et al. Cryo-EM structure of oxysterol-bound human smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283 (2019).
Nusse, R. & Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Hirai, H. et al. Crystal structure of a mammalian Wnt-frizzled complex. Nat. Struct. Mol. Biol. 26, 372–379 (2019).
Shen, G. et al. Structural basis of the Norrin-Frizzled 4 interaction. Cell Res. 25, 1078–1081 (2015).
Huang, P. et al. Cellular cholesterol directly activates smoothened in Hedgehog signaling. Cell 166, 1176.e14–1187.e14 (2016).
Anighoro, A., Bajorath, J. & Rastelli, G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 57, 7874–7887 (2014).
Corbett, A., Williams, G. & Ballard, C. Drug repositioning: an opportunity to develop novel treatments for Alzheimer’s disease. Pharmaceuticals 6, 1304–1321 (2013).
Ravikumar, B. & Aittokallio, T. Improving the efficacy-safety balance of polypharmacology in multi-target drug discovery. Expert Opin. Drug Discov. 13, 179–192 (2018).
Oprea, T. I. et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 17, 317–332 (2018).
Palacios, J. M., Pazos, A. & Hoyer, D. A short history of the 5-HT2C receptor: from the choroid plexus to depression, obesity and addiction treatment. Psychopharmacology 234, 1395–1418 (2017).
Pogorelov, V. M. et al. 5-HT2C agonists modulate schizophrenia-like behaviors in mice. Neuropsychopharmacology 42, 2163–2177 (2017).
McCorvy, J. D. & Roth, B. L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 150, 129–142 (2015).
Sexton, P. M. & Christopoulos, A. To bind or not to bind: unravelling GPCR polypharmacology. Cell 172, 636–638 (2018).
Garland, S. L. Are GPCRs still a source of new targets? J. Biomol. Screen. 18, 947–966 (2013).
Yang, P. Y. et al. Stapled, long-acting glucagon-like peptide 2 analog with efficacy in dextran sodium sulfate induced mouse colitis models. J. Med. Chem. 61, 3218–3223 (2018).
Nichols, D. E. Psychedelics. Pharmacol. Rev. 68, 264–355 (2016).
Butini, S. et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 142, 68–103 (2016).
Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017).
Wu, Z. et al. Quantitative and systems pharmacology 2. In silico polypharmacology of G protein-coupled receptor ligands via network-based approaches. Pharmacol. Res. 129, 400–413 (2018).
Huang, X. P. et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527, 477–483 (2015).
Jacoby, E., Bouhelal, R., Gerspacher, M. & Seuwen, K. The 7 TM G-protein-coupled receptor target family. ChemMedChem 1, 761–782 (2006).
Quinones, M. et al. Exciting advances in GPCR-based drugs discovery for treating metabolic disease and future perspectives. Expert Opin. Drug Discov. 14, 421–431 (2019).
Smith, J. S., Lefkowitz, R. J. & Rajagopal, S. Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 17, 243–260 (2018).
Tan, L., Yan, W., McCorvy, J. D. & Cheng, J. Biased ligands of G protein-coupled receptors (GPCRs): structure-functional selectivity relationships (SFSRs) and therapeutic potential. J. Med. Chem. 61, 9841–9878 (2018).
Ok, H. G. et al. Can oliceridine (TRV130), an ideal novel micro receptor G protein pathway selective (micro-GPS) modulator, provide analgesia without opioid-related adverse reactions? Korean J. Pain 31, 73–79 (2018).
Zebala, J. A., Schuler, A. D., Kahn, S. J. & Maeda, D. Y. Desmetramadol is identified as a G-protein biased micro opioid receptor agonist. Front. Pharmacol. 10, 1680 (2019).
Bedini, A. et al. Functional selectivity and antinociceptive effects of a novel KOPr agonist. Front. Pharmacol. 11, 188 (2020).
James, I. E. et al. A first-in-human clinical study with TRV734, an orally bioavailable G-protein-biased ligand at the mu-opioid receptor. Clin. Pharmacol. Drug Dev. 9, 256–266 (2020).
Hill, R. et al. The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 175, 2653–2661 (2018).
Ehrlich, A. T. et al. Biased signaling of the mu opioid receptor revealed in native neurons. iScience 14, 47–57 (2019).
White, K. L. et al. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 352, 98–109 (2015).
Mores, K. L., Cummins, B. R., Cassell, R. J. & van Rijn, R. M. A review of the therapeutic potential of recently developed G protein-biased kappa agonists. Front. Pharmacol. 10, 407 (2019).
Kozono, H., Yoshitani, H. & Nakano, R. Post-marketing surveillance study of the safety and efficacy of nalfurafine hydrochloride (Remitch® capsules 2.5 μg) in 3,762 hemodialysis patients with intractable pruritus. Int. J. Nephrol. Renovasc. Dis. 11, 9–24 (2018).
Okeke, K., Michel-Reher, M. B., Gravas, S. & Michel, M. C. Desensitization of cAMP accumulation via human beta3-adrenoceptors expressed in human embryonic kidney cells by full, partial, and biased agonists. Front. Pharmacol. 10, 596 (2019).
Cernecka, H., Sand, C. & Michel, M. C. The odd sibling: features of beta3-adrenoceptor pharmacology. Mol. Pharmacol. 86, 479–484 (2014).
Baker, J. G., Hill, S. J. & Summers, R. J. Evolution of beta-blockers: from anti-anginal drugs to ligand-directed signalling. Trends Pharmacol. Sci. 32, 227–234 (2011).
Xiao, C., Goldgof, M., Gavrilova, O. & Reitman, M. L. Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity 23, 1450–1459 (2015).
Lei, X. & Wong, G. W. C1q/TNF-related protein 2 (CTRP2) deletion promotes adipose tissue lipolysis and hepatic triglyceride secretion. J. Biol. Chem. 294, 15638–15649 (2019).
Hoare, S. R. J., Tewson, P. H., Quinn, A. M. & Hughes, T. E. A kinetic method for measuring agonist efficacy and ligand bias using high resolution biosensors and a kinetic data analysis framework. Sci. Rep. 10, 1766 (2020).
Pang, P. S. et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: a randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Heart J. 38, 2364–2373 (2017).
Namkung, Y. et al. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal. 11, eaat1631 (2018).
Schena, G. & Caplan, M. J. Everything you always wanted to know about beta3-AR * (* but were afraid to ask). Cells 8, 357 (2019).
Schattauer, S. S., Kuhar, J. R., Song, A. & Chavkin, C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal. 32, 59–65 (2017).
Lindsley, C. W. et al. Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chem. Rev. 116, 6707–6741 (2016).
Congreve, M., Oswald, C. & Marshall, F. H. Applying structure-based drug design approaches to allosteric modulators of GPCRs. Trends Pharmacol. Sci. 38, 837–847 (2017).
Wold, E. A., Chen, J., Cunningham, K. A. & Zhou, J. Allosteric modulation of class A GPCRs: targets, agents, and emerging concepts. J. Med. Chem. 62, 88–127 (2019).
Wu, Y. et al. GPCR allosteric modulator discovery. Adv. Exp. Med. Biol. 1163, 225–251 (2019).
Liu, X. et al. Unraveling allosteric landscapes of allosterome with ASD. Nucleic Acids Res. 48, D394–D401 (2020).
Nolte, W. M. et al. A potentiator of orthosteric ligand activity at GLP-1R acts via covalent modification. Nat. Chem. Biol. 10, 629–631 (2014).
Bock, A., Schrage, R. & Mohr, K. Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 136, 427–437 (2018).
Price, M. R. et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 68, 1484–1495 (2005).
Shao, Z. et al. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 15, 1199–1205 (2019).
Goupil, E. et al. A novel biased allosteric compound inhibitor of parturition selectively impedes the prostaglandin F2alpha-mediated Rho/ROCK signaling pathway. J. Biol. Chem. 285, 25624–25636 (2010).
Quoyer, J. et al. Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc. Natl Acad. Sci. USA 110, E5088–E5097 (2013).
Zhang, D. et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520, 317–321 (2015).
Brodbeck, R. M. et al. al. Identification and characterization of NDT 9513727 [N,N-bis(1,3-benzodioxol-5-ylmethyl)-1-butyl-2,4-diphenyl-1H-imidazole-5-methanamine], a novel, orally bioavailable C5a receptor inverse agonist. J. Pharmacol. Exp. Ther 327, 898–909 (2008).
Robertson, N. et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114 (2018).
Maeda, S. et al. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364, 552–557 (2019).
Staus, D. P. et al. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 579, 297–302 (2020).
Shore, D. M. et al. Allosteric modulation of a cannabinoid G protein-coupled receptor: binding site elucidation and relationship to G protein signaling. J. Biol. Chem. 289, 5828–5845 (2014).
Stornaiuolo, M. et al. Endogenous vs exogenous allosteric modulators in GPCRs: a dispute for shuttling CB1 among different membrane microenvironments. Sci. Rep. 5, 15453 (2015).
Fay, J. F. & Farrens, D. L. The membrane proximal region of the cannabinoid receptor CB1 N-terminus can allosterically modulate ligand affinity. Biochemistry 52, 8286–8294 (2013).
Ahn, K. H., Mahmoud, M. M. & Kendall, D. A. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J. Biol. Chem. 287, 12070–12082 (2012).
Baillie, G. L. et al. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol. Pharmacol. 83, 322–338 (2013).
Hua, T. et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547, 468–471 (2017).
Liu, X. et al. An allosteric modulator binds to a conformational hub in the beta2 adrenergic receptor. Nat. Chem. Biol. 16, 749–755 (2020).
Liu, X. et al. Mechanism of beta2AR regulation by an intracellular positive allosteric modulator. Science 364, 1283–1287 (2019).
Liu, H. et al. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 25, 472–481 (2018).
Glukhova, A. et al. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 168, 867–877 e813 (2017).
Ho, J. D. et al. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun. 9, 1645 (2018).
Wu, F. et al. Full-length human GLP-1 receptor structure without orthosteric ligands. Nat. Commun. 11, 1272 (2020).
Muniz-Medina, V. M. et al. The relative activity of "function sparing" HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol. Pharmacol. 75, 490–501 (2009).
Tan, Q. et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 (2013).
Zheng, Y. et al. Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46, 1005–1017 e1005 (2017).
Shaik, M. M. et al. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 565, 318–323 (2019).
Zhao, P. et al. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 577, 432–436 (2020).
Zhang, Y. et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017).
Zheng, Y. et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540, 458–461 (2016).
Jaeger, K. et al. Structural basis for allosteric ligand recognition in the human CC chemokine receptor 7. Cell 178, 1222.e10–1230.e10 (2019).
Oswald, C. et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature 540, 462–465 (2016).
Liu, X. et al. Mechanism of intracellular allosteric beta2AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484 (2017).
Wingler, L. M. et al. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 176, 479.e12–490.e12 (2019).
Wingler, L. M. et al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367, 888–892 (2020).
Lee, Y. et al. Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 583, 862–866 (2020).
Leach, K. & Gregory, K. J. Molecular insights into allosteric modulation of Class C G protein-coupled receptors. Pharmacol. Res. 116, 105–118 (2017).
Weierstall, U. et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 (2014).
Wang, C. et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 (2013).
Crowther, G. J. et al. Cofactor-independent phosphoglycerate mutase from nematodes has limited druggability, as revealed by two high-throughput screens. PLoS Negl. Trop. Dis. 8, e2628 (2014).
Zhou, F. et al. Colocalization strategy unveils an underside binding site in the transmembrane domain of smoothened receptor. J. Med. Chem. 62, 9983–9989 (2019).
Huang, P. et al. Structural basis of smoothened activation in Hedgehog signaling. Cell 174, 312.e16–324.e16 (2018).
Yu, W. & MacKerell, A. D. Jr. Computer-aided drug design methods. Methods Mol. Biol. 1520, 85–106 (2017).
Caffrey, M. & Cherezov, V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731 (2009).
Erlandson, S. C., McMahon, C. & Kruse, A. C. Structural basis for G protein-coupled receptor signaling. Annu. Rev. Biophys. 47, 1–18 (2018).
Lyu, J. et al. Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019).
Stein, R. M. et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 579, 609–614 (2020).
Ballante, F. et al. Docking finds GPCR ligands in dark chemical matter. J. Med. Chem. 63, 613–620 (2020).
Ranganathan, A. et al. Ligand discovery for a peptide-binding GPCR by structure-based screening of fragment- and lead-like chemical libraries. ACS Chem. Biol. 12, 735–745 (2017).
Liu, X. et al. Salvianolic acids from antithrombotic Traditional Chinese Medicine Danshen are antagonists of human P2Y1 and P2Y12 receptors. Sci. Rep. 8, 8084 (2018).
Bissantz, C., Schalon, C., Guba, W. & Stahl, M. Focused library design in GPCR projects on the example of 5-HT(2c) agonists: comparison of structure-based virtual screening with ligand-based search methods. Proteins 61, 938–952 (2005).
Mannel, B. et al. Structure-guided screening for functionally selective D2 dopamine receptor ligands from a virtual chemical library. ACS Chem. Biol. 12, 2652–2661 (2017).
Guo, T. & Hobbs, D. W. Privileged structure-based combinatorial libraries targeting G protein-coupled receptors. Assay Drug Dev. Technol. 1, 579–592 (2003).
Latorraca, N. R., Venkatakrishnan, A. J. & Dror, R. O. GPCR dynamics: structures in motion. Chem. Rev. 117, 139–155 (2017).
Lee, Y., Lazim, R., Macalino, S. J. Y. & Choi, S. Importance of protein dynamics in the structure-based drug discovery of class A G protein-coupled receptors (GPCRs). Curr. Opin. Struct. Biol. 55, 147–153 (2019).
Hilger, D., Masureel, M. & Kobilka, B. K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 (2018).
Vilar, S. & Costanzi, S. In G Protein Coupled Receptors: Modeling, Activation, Interactions and Virtual Screening. Methods in Enzymology, Vol. 522 (ed. Conn P. M.) 263–278 (Elsevier Academic Press, 2013).
Coudrat, T., Christopoulos, A., Sexton, P. M. & Wootten, D. Structural features embedded in G protein-coupled receptor co-crystal structures are key to their success in virtual screening. PLoS ONE 12, e0174719 (2017).
Miao, Y. et al. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc. Natl Acad. Sci. USA 113, E5675–5684 (2016).
Warszycki, D. et al. From homology models to a set of predictive binding pockets-a 5-HT1A receptor case study. J. Chem. Inf. Model. 57, 311–321 (2017).
de Graaf, C. et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J. Med. Chem. 54, 8195–8206 (2011).
David, L., Nielsen, P. A., Hedstrom, M. & Norden, B. Scope and limitation of ligand docking: methods, scoring functions and protein targets. Curr. Comput. Aided Drug Des. 1, 275–306 (2005).
Kooistra, A. J. et al. Function-specific virtual screening for GPCR ligands using a combined scoring method. Sci. Rep. 6, 28288 (2016).
Bartuzi, D., Kaczor, A. A., Targowska-Duda, K. M. & Matosiuk, D. Recent advances and applications of molecular docking to G protein-coupled receptors. Molecules 22, 23 (2017).
Zhou, Y. et al. Structure-based discovery of novel and selective 5-hydroxytryptamine 2B receptor antagonists for the treatment of irritable bowel syndrome. J. Med. Chem. 59, 707–720 (2016).
Rastelli, G. & Pinzi, L. Refinement and rescoring of virtual screening results. Front. Chem. 7, 498 (2019).
Lenselink, E. B. et al. Predicting binding affinities for GPCR ligands using free-energy perturbation. ACS Omega 1, 293–304 (2016).
Kim, M. & Cho, A. E. Incorporating QM and solvation into docking for applications to GPCR targets. Phys. Chem. Chem. Phys. 18, 28281–28289 (2016).
Heifetz, A. et al. Using the fragment molecular orbital method to investigate agonist-orexin-2 receptor interactions. Biochem. Soc. Trans. 44, 574–581 (2016).
Heifetz, A. et al. The fragment molecular orbital method reveals new insight into the chemical nature of GPCR-ligand interactions. J. Chem. Inf. Model. 56, 159–172 (2016).
Zhou, Q. et al. Exploring the mutational robustness of nucleic acids by searching genotype neighborhoods in sequence space. J. Phys. Chem. Lett. 8, 407–414 (2017).
Hou, T., Wang, J., Li, Y. & Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82 (2011).
Yau, M. Q. et al. Evaluating the performance of MM/PBSA for binding affinity prediction using class A GPCR crystal structures. J. Comput. Aided Mol. Des. 33, 487–496 (2019).
Kooistra, A. J., Leurs, R., de Esch, I. J. & de Graaf, C. Structure-based prediction of G-protein-coupled receptor ligand function: a beta-adrenoceptor case study. J. Chem. Inf. Model. 55, 1045–1061 (2015).
Fan, L. et al. Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands. Nat. Commun. 11, 1074 (2020).
Kruse, A. C. et al. Muscarinic receptors as model targets and antitargets for structure-based ligand discovery. Mol. Pharmacol. 84, 528–540 (2013).
Wei, Y. et al. Identification of new potent A1 adenosine receptor antagonists using a multistage virtual screening approach. Eur. J. Med. Chem. 187, 111936 (2020).
Suomivuori, C. M. et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 367, 881–887 (2020).
McCorvy, J. D. et al. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 14, 126–134 (2018).
Lu, S. & Zhang, J. Small molecule allosteric modulators of G-protein-coupled receptors: drug-target interactions. J. Med. Chem. 62, 24–45 (2019).
Luckmann, M. et al. Molecular dynamics-guided discovery of an ago-allosteric modulator for GPR40/FFAR1. Proc. Natl Acad. Sci. USA 116, 7123–7128 (2019).
Weis, W. I. & Kobilka, B. K. The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018).
Raschka, S. Automated discovery of GPCR bioactive ligands. Curr. Opin. Struct. Biol. 55, 17–24 (2019).
Kumari, P., Ghosh, E. & Shukla, A. K. Emerging approaches to GPCR ligand screening for drug discovery. Trends Mol. Med. 21, 687–701 (2015).
Chen, L., Jin, L. & Zhou, N. An update of novel screening methods for GPCR in drug discovery. Expert Opin. Drug Discov. 7, 791–806 (2012).
Neri, D. & Lerner, R. A. DNA-encoded chemical libraries: a selection system based on endowing organic compounds with amplifiable information. Annu. Rev. Biochem. 87, 479–502 (2018).
Kleiner, R. E., Dumelin, C. E. & Liu, D. R. Small-molecule discovery from DNA-encoded chemical libraries. Chem. Soc. Rev. 40, 5707–5717 (2011).
Goodnow, R. A. Jr., Dumelin, C. E. & Keefe, A. D. DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 16, 131–147 (2017).
Kodadek, T., Paciaroni, N. G., Balzarini, M. & Dickson, P. Beyond protein binding: recent advances in screening DNA-encoded libraries. Chem. Commun. 55, 13330–13341 (2019).
Ahn, S. et al. Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc. Natl Acad. Sci. USA 114, 1708–1713 (2017).
Ahn, S. et al. Small-molecule positive allosteric modulators of the beta2-adrenoceptor isolated from DNA-encoded libraries. Mol. Pharmacol. 94, 850–861 (2018).
Brown, D. G. et al. Agonists and antagonists of protease-activated receptor 2 discovered within a DNA-encoded chemical library using mutational stabilization of the target. SLAS Discov. 23, 429–436 (2018).
Wu, Z. et al. Cell-based selection expands the utility of DNA-encoded small-molecule library technology to cell surface drug targets: identification of novel antagonists of the NK3 tachykinin receptor. ACS Comb. Sci. 17, 722–731 (2015).
Annis, A., Chuang, C. C. & Nazef, N. In Mass Spectrometry in Medicinal Chemistry: Applications in Drug Discovery. Methods and Principles in Medicinal Chemistry (eds Wanner, K. T. & Höfner, G.) 121–156 (Wiley, 2007).
O’Connell, T. N. et al. Solution-based indirect affinity selection mass spectrometry–a general tool for high-throughput screening of pharmaceutical compound libraries. Anal. Chem. 86, 7413–7420 (2014).
Chen, X. et al. Identification of inhibitors of the antibiotic-resistance target New Delhi metallo-beta-lactamase 1 by both nanoelectrospray ionization mass spectrometry and ultrafiltration liquid chromatography/mass spectrometry approaches. Anal. Chem. 85, 7957–7965 (2013).
Chen, X. et al. A ligand-observed mass spectrometry approach integrated into the fragment based lead discovery pipeline. Sci. Rep. 5, 8361 (2015).
Qin, S. et al. Multiple ligand detection and affinity measurement by ultrafiltration and mass spectrometry analysis applied to fragment mixture screening. Anal. Chim. Acta 886, 98–106 (2015).
Gesmundo, N. J. et al. Nanoscale synthesis and affinity ranking. Nature 557, 228–232 (2018).
Whitehurst, C. E. et al. Application of affinity selection-mass spectrometry assays to purification and affinity-based screening of the chemokine receptor CXCR4. Comb. Chem. High Throughput Screen. 15, 473–485 (2012).
Ma, J. et al. Ligand identification of the adenosine A2A receptor in self-assembled nanodiscs by affinity mass spectrometry. Anal. Methods 9, 5851–5858 (2017).
Calleri, E. et al. Frontal affinity chromatography-mass spectrometry useful for characterization of new ligands for GPR17 receptor. J. Med. Chem. 53, 3489–3501 (2010).
Temporini, C. et al. Development of new chromatographic tools based on A2A adenosine receptor subtype for ligand characterization and screening by FAC-MS. Anal. Bioanal. Chem. 405, 837–845 (2013).
Qin, S. et al. High-throughput identification of G protein-coupled receptor modulators through affinity mass spectrometry screening. Chem. Sci. 9, 3192–3199 (2018).
Massink, A. et al. Mass spectrometry-based ligand binding assays on adenosine A1 and A2A receptors. Purinergic Signal. 11, 581–594 (2015).
Yen, H. Y. et al. Ligand binding to a G protein-coupled receptor captured in a mass spectrometer. Sci. Adv. 3, e1701016 (2017).
Yen, H. Y. et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 423–427 (2018).
Deng, Y. et al. Discovery of novel, dual mechanism ERK inhibitors by affinity selection screening of an inactive kinase. J. Med. Chem. 57, 8817–8826 (2014).
Zhang, T. et al. Definitive metabolite identification coupled with automated ligand identification system (ALIS) technology: a novel approach to uncover structure-activity relationships and guide drug design in a factor IXa inhibitor program. J. Med. Chem. 59, 1818–1829 (2016).
Kutilek, V. D. et al. Integration of affinity selection-mass spectrometry and functional cell-based assays to rapidly triage druggable target space within the NF-kappaB pathway. J. Biomol. Screen. 21, 608–619 (2016).
Walker, S. S. et al. Affinity selection-mass spectrometry identifies a novel antibacterial RNA polymerase inhibitor. ACS Chem. Biol. 12, 1346–1352 (2017).
Whitehurst, C. E. et al. Discovery and characterization of orthosteric and allosteric muscarinic M2 acetylcholine receptor ligands by affinity selection-mass spectrometry. J. Biomol. Screen. 11, 194–207 (2006).
Lu, Y. et al. Accelerating the throughput of affinity mass spectrometry-based ligand screening toward a G protein-coupled receptor. Anal. Chem. 91, 8162–8169 (2019).
Choi, Y. et al. Screening natural products for inhibitors of quinone reductase-2 using ultrafiltration LC-MS. Anal. Chem. 83, 1048–1052 (2011).
Yang, Z. et al. An ultrafiltration high-performance liquid chromatography coupled with diode array detector and mass spectrometry approach for screening and characterising tyrosinase inhibitors from mulberry leaves. Anal. Chim. Acta 719, 87–95 (2012).
Song, H. P. et al. A strategy for screening of high-quality enzyme inhibitors from herbal medicines based on ultrafiltration LC-MS and in silico molecular docking. Chem. Commun. 51, 1494–1497 (2015).
Fu, X. et al. Novel chemical ligands to ebola virus and marburg virus nucleoproteins identified by combining affinity mass spectrometry and metabolomics approaches. Sci. Rep. 6, 29680 (2016).
Wang, L. et al. Quickly screening for potential alpha-glucosidase inhibitors from guava leaves tea by bioaffinity ultrafiltration coupled with HPLC-ESI-TOF/MS method. J. Agric. Food Chem. 66, 1576–1582 (2018).
Wang, Z. et al. Efficient ligand discovery from natural herbs by integrating virtual screening, affinity mass spectrometry and targeted metabolomics. Analyst 144, 2881–2890 (2019).
Zhang, B. et al. A novel G protein-biased and subtype-selective agonist for a G protein-coupled receptor discovered from screening herbal extracts. ACS Cent. Sci. 6, 213–225 (2020).
Udugamasooriya, D. G., Dineen, S. P., Brekken, R. A. & Kodadek, T. A peptoid “antibody surrogate” that antagonizes VEGF receptor 2 activity. J. Am. Chem. Soc. 130, 5744–5752 (2008).
Hofner, G. & Wanner, K. T. Competitive binding assays made easy with a native marker and mass spectrometric quantification. Angew. Chem. Int. Ed. 42, 5235–5237 (2003).
Niessen, K. V., Hofner, G. & Wanner, K. T. Competitive MS binding assays for dopamine D2 receptors employing spiperone as a native marker. ChemBioChem 6, 1769–1775 (2005).
Zepperitz, C., Hofner, G. & Wanner, K. T. MS-binding assays: kinetic, saturation, and competitive experiments based on quantitation of bound marker as exemplified by the GABA transporter mGAT1. ChemMedChem 1, 208–217 (2006).
Grimm, S. H., Hofner, G. & Wanner, K. T. MS binding assays for the three monoamine transporters using the triple reuptake inhibitor (1R,3S)-indatraline as native marker. ChemMedChem 10, 1027–1039 (2015).
Kern, F. T. & Wanner, K. T. Generation and screening of oxime libraries addressing the neuronal GABA transporter GAT1. ChemMedChem 10, 396–410 (2015).
Sichler, S. et al. Development of MS binding assays targeting the binding site of MB327 at the nicotinic acetylcholine receptor. Toxicol. Lett. 293, 172–183 (2018).
Neiens, P., Hofner, G. & Wanner, K. T. MS binding assays for D1 and D5 dopamine receptors. ChemMedChem 10, 1924–1931 (2015).
Schuller, M., Hofner, G. & Wanner, K. T. Simultaneous multiple MS binding assays addressing D1 and D2 dopamine receptors. ChemMedChem 12, 1585–1594 (2017).
Sanchez-Garrido, M. A. et al. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 60, 1851–1861 (2017).
Brandt, S. J., Gotz, A., Tschop, M. H. & Muller, T. D. Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides 100, 190–201 (2018).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Zhou, F. et al. Structural basis for activation of the growth hormone-releasing hormone receptor. Nat. Commun. 11, 5205 (2020).
Sun, W. et al. A unique hormonal recognition feature of the human glucagon-like peptide-2 receptor. Cell Res. 30, 1098–1108 (2020).
Seyedabadi, M., Ghahremani, M. H. & Albert, P. R. Biased signaling of G protein coupled receptors (GPCRs): molecular determinants of GPCR/transducer selectivity and therapeutic potential. Pharmacol. Ther. 200, 148–178 (2019).
Cussac, D. et al. Agonist-directed trafficking of signalling at serotonin 5-HT2A, 5-HT2B and 5-HT2C-VSV receptors mediated Gq/11 activation and calcium mobilisation in CHO cells. Eur. J. Pharmacol. 594, 32–38 (2008).
Brust, T. F. et al. Bias analyses of preclinical and clinical D2 dopamine ligands: studies with immediate and complex signaling pathways. J. Pharmacol. Exp. Ther. 352, 480–493 (2015).
Wacker, D. et al. Structural features for functional selectivity at serotonin receptors. Science 340, 615–619 (2013).
Stewart, G. D., Sexton, P. M. & Christopoulos, A. Detection of novel functional selectivity at M3 muscarinic acetylcholine receptors using a Saccharomyces cerevisiae platform. ACS Chem. Biol. 5, 365–375 (2010).
Pronin, A. N., Wang, Q. & Slepak, V. Z. Teaching an old drug new tricks: agonism, antagonism, and biased signaling of pilocarpine through M3 muscarinic acetylcholine receptor. Mol. Pharmacol. 92, 601–612 (2017).
Baltos, J. A. et al. Quantification of adenosine A(1) receptor biased agonism: implications for drug discovery. Biochem. Pharmacol. 99, 101–112 (2016).
Hodavance, S. Y., Gareri, C., Torok, R. D. & Rockman, H. A. G Protein-coupled receptor biased agonism. J. Cardiovasc. Pharmacol. 67, 193–202 (2016).
Drake, M. T. et al. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J. Biol. Chem. 283, 5669–5676 (2008).
Yano, H. et al. Gs- versus Golf-dependent functional selectivity mediated by the dopamine D1 receptor. Nat. Commun. 9, 486 (2018).
Klein Herenbrink, C. et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 7, 10842 (2016).
Webster, L. & Schmidt, W. K. Dilemma of addiction and respiratory depression in the treatment of pain: a prototypical endomorphin as a new approach. Pain Med. 21, 992–1004 (2020).
Rahmeh, R. et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl Acad. Sci. USA 109, 6733–6738 (2012).
Busnelli, M. et al. Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J. Biol. Chem. 287, 3617–3629 (2012).
Dhar, T. G. et al. Identification and preclinical pharmacology of BMS-986104: a differentiated S1P1 receptor modulator in clinical trials. ACS Med. Chem. Lett. 7, 283–288 (2016).
Lin, X. et al. Slowly signaling G protein-biased CB2 cannabinoid receptor agonist ly2828360 suppresses neuropathic pain with sustained efficacy and attenuates morphine tolerance and dependence. Mol. Pharmacol. 93, 49–62 (2018).
Boatman, P. D. et al. (1aR,5aR)1a,3,5,5a-Tetrahydro-1H-2,3-diaza-cyclopropa[a]pentalene-4-carboxylic acid (MK-1903): a potent GPR109a agonist that lowers free fatty acids in humans. J. Med. Chem. 55, 3644–3666 (2012).
Cruz-Monteagudo, M. et al. Systemic QSAR and phenotypic virtual screening: chasing butterflies in drug discovery. Drug Discov. Today 22, 994–1007 (2017).
Fan, F., Toledo Warshaviak, D., Hamadeh, H. K. & Dunn, R. T. II The integration of pharmacophore-based 3D QSAR modeling and virtual screening in safety profiling: a case study to identify antagonistic activities against adenosine receptor, A2A, using 1,897 known drugs. PLoS ONE 14, e0204378 (2019).
von Korff, M. & Steger, M. GPCR-tailored pharmacophore pattern recognition of small molecular ligands. J. Chem. Inf. Comput Sci. 44, 1137–1147 (2004).
He, G. et al. An improved receptor-based pharmacophore generation algorithm guided by atomic chemical characteristics and hybridization types. Front Pharmacol. 9, 1463 (2018).
Acknowledgements
The authors acknowledge funding support from the National Natural Science Foundation of China 81872915 (to M.-W.W.), 81773792 (to D.Y.), 81973373 (to D.Y.), 21704064 (to Q.Z.), 31971362 (to W.S.), 31971178 (to S.Z.), and 31770796 (to Y.J.); National Science & Technology Major Project of China – Key New Drug Creation and Manufacturing Program 2018ZX09735-001 (to M.-W.W.), 2018ZX09711002-002-005 (to D.Y.), and 2018ZX09711002-002-002 (to Y.J.); the National Key Basic Research Program of China 2018YFA0507000 (to M.-W.W., S.Z., W.S., and H.T.); Novo Nordisk-CAS Research Fund grant NNCAS-2017-1-CC (to D.Y.); The Belt and Road Master Fellowship program (to V.L.); UCAS Scholarship for International Students (to S.D.); and The CAS-TWAS President’s Fellowship for International Doctoral Students (to E.Y.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, D., Zhou, Q., Labroska, V. et al. G protein-coupled receptors: structure- and function-based drug discovery. Sig Transduct Target Ther 6, 7 (2021). https://doi.org/10.1038/s41392-020-00435-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00435-w
This article is cited by
-
Long noncoding RNAs and mRNAs profiling in ovary during laying and broodiness in Taihe Black-Bone Silky Fowls (Gallus gallus Domesticus Brisson)
BMC Genomics (2024)
-
G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery
Signal Transduction and Targeted Therapy (2024)
-
Cryo-electron microscopy for GPCR research and drug discovery in endocrinology and metabolism
Nature Reviews Endocrinology (2024)
-
Emerging mechanistic understanding of cilia function in cellular signalling
Nature Reviews Molecular Cell Biology (2024)
-
First report on chemometrics-driven multilayered lead prioritization in addressing oxysterol-mediated overexpression of G protein-coupled receptor 183
Molecular Diversity (2024)
