The catastrophic global health and socioeconomic impact of COVID-19, together with the absence of any clearly effective preventive or therapeutic remedies, has created a massive unmet medical need. Rapid responses by governments, academia and industry have already resulted in the production of more than 180 vaccine candidates1, 42 of which are being tested in humans at the time of writing. The considerable design flexibility of newer types of vaccine technology gave these candidates a head start in the race. Some of the candidates, which are based on nucleic acids (such as messenger RNA), entered human trials2 as early as March. In this issue, Mulligan et al.3 and Sahin et al.4 report clinical-trial results for a COVID-19 vaccine called BNT162b1, which contains mRNA that encodes part of a protein found on the surface of the SARS-CoV-2 coronavirus. This vaccine, made by Pfizer and BioNTech, was tested in adults in a combined phase I and phase II clinical trial.
Read the paper: SARS-CoV-2 vaccines in development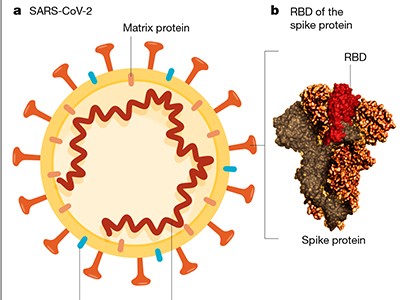
The primary goal of phase I/II vaccine clinical trials is to evaluate short-term safety, check dosage and assess aspects of the body’s reaction to the vaccine — effects known as reactogenicity. Reactogenicity could include localized pain, redness or swelling at the site of vaccine injection, as well as systemic symptoms elsewhere in the body, such as fever, muscle pain and headache. Some reactogenicity might be expected as a normal sign that the immune system is generating a response to the vaccine, and so early-phase safety evaluation focuses particularly on more-serious effects.
The secondary goal of these early-stage clinical trials is to assess immunogenicity — the ability of a vaccine to stimulate a detectable immune response to the vaccine target (Fig. 1). This typically involves assessing components of what is known as the adaptive branch of the immune system. The features of interest are vaccine-specific antibody responses and immune cells called CD4 (or helper) T cells and CD8 (cytotoxic) T cells. These T cells can directly target cells infected with the virus, or collaborate with antibody-producing B cells. The best vaccines elicit long-lasting responses that produce ‘neutralizing’ antibodies, which act to hinder or prevent an infectious agent from causing illness5. Once phase I and II trials have been completed, a phase III study can be conducted to determine whether the vaccine affects how susceptible people are to a disease.

Figure 1 | Assessing a vaccine that targets the SARS-CoV-2 coronavirus. Mulligan et al.3 and Sahin et al.4 report vaccine clinical-trial results for a combined phase I and phase II study. a, The vaccine contains messenger RNA in a lipid nanoparticle that is taken up by human cells. The mRNA encodes a region of the coronavirus spike protein termed the receptor-binding domain (RBD). b, After vaccination, the authors monitored the trial participants’ responses. One type of response assessed was ‘reactogenicity’ — signs of the body’s reaction to vaccination, such as local swelling at the injection site or systemic effects elsewhere in the body, for example, headache or fever. Another type of response assessed, called immunogenicity, relates to signs of an immune-system defence against the vaccine target, as indicated by the presence of RBD-specific antibodies, and RBD-specific T cells that produce the signalling molecule interferon-γ. The reactogenicity and immunogenicity results were acceptable for this early-stage clinical-trial work.
Mulligan, Sahin and their respective colleagues provide the first insights into the reactogenicity and immunogenicity of BNT162b1. This vaccine consists of an injected mRNA that encodes part of the ‘spike’ protein of SARS-CoV-2 — a region of the protein known as the receptor-binding domain (RBD), which enables the virus to engage with and infect human cells. Antibodies that bind to the RBD provide a way of interfering with a key starting point in the SARS-CoV-2 cycle of infection, and so attack this viral Achilles heel. Accordingly, the RBD and the spike protein are the targets of most of the vaccine candidates.
Read the paper: Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults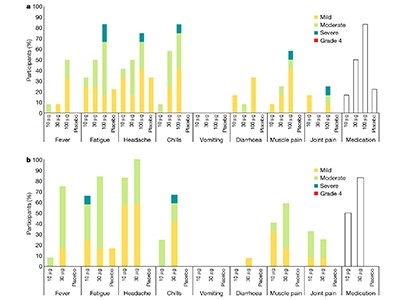
Mulligan and colleagues gave the vaccine at one of 3 doses (10, 30 and 100 micrograms) to 36 healthy adults (age range 18–55 years), with 9 other participants receiving a placebo treatment. Sahin and colleagues’ trial did not have a placebo control group, and enrolled 60 participants who received the vaccine at one of 5 doses (1, 10, 30, 50 and 60 µg). The participants in both trials in all but the highest dosage groups (100 and 60 µg, respectively) received 2 vaccinations at 3-week intervals, in what is known as a prime–boost regimen. This approach can determine whether the addition of a second ‘booster’ vaccination enables a strong immune response to develop. More than 80% of the study participants were white, and around 2% were Black.
Although no serious adverse events were reported, notable reactions at the injection site or elsewhere in the body were frequent. For example, of the participants in the medium-dose (30 µg) group of both studies, 96% reported pain at the injection site and 92% reported headaches. Moreover, the prevalence of these reactions was dose dependent, and increased after the booster immunization, so a second injection was not given to the highest-dose groups. In addition, lymphocytes — white blood cells of the immune system (which include T cells and B cells) — were reduced in number in most vaccinated individuals, but returned to normal 6–8 days after vaccination.
Vaccine-induced anti-RBD antibody levels were quantified at multiple time points. However, the latest time point assessed was at only two (Mulligan et al.) or three (Sahin et al.) weeks after the booster injection. All vaccinees developed low-level anti-RBD antibody responses after the first vaccination. As expected, the antibody levels depended on the vaccine dose, and they increased 10–15-fold after the booster. By three weeks after the booster, the antibody levels dropped. Antibody-mediated SARS-CoV-2 neutralization, as assessed by in vitro experiments, followed a similar pattern, and it also declined three weeks after the booster. This result stresses the importance of long-term follow-up to understand the durability of vaccine-induced immune responses. A decline in the response is expected over time, and such a follow-up is needed to determine the rapidity of this decline.
With the exception of the group who received the lowest vaccine dose, levels of neutralizing-antibodies against SARS-CoV-2 compared favourably with those in blood samples taken from people who had recovered from COVID-19 — commonly referred to as COVID-19 convalescent serum or plasma. Crucially, the magnitude and dynamics of the elicited antibody response indicate that a booster dose is essential for this vaccine.
Read the paper: COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses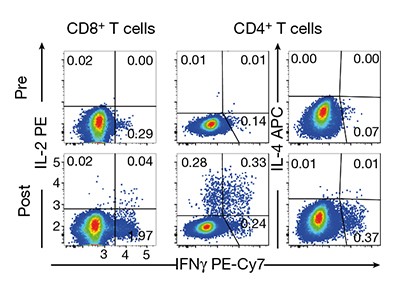
Sahin and colleagues measured the responses of CD4 and CD8 T cells before the first vaccination and one week after the booster. Although most vaccinees showed convincing responses, the strength of the T-cell responses, as measured by the production of immune-system signalling molecules called cytokines, varied between participants, and there was no clear dose dependency in the responses.
In terms of what we have learnt from the results of these phase I/II clinical trials, the reactogenicity and early safety profile seem acceptable. However, it should be remembered that, as the authors acknowledge, this was a small group of individuals, and it was missing people from key age profiles and at-risk groups. The average age of the participants in the two trials was 35 and 37, respectively.
In another study6, Pfizer and BioNTech reported a clinical trial that compared BNT162b1 with a different version of the vaccine, termed BNT162b2, that uses mRNA encoding the full-length spike protein. Among older adults, aged between 65 and 85, those vaccinated with BNT162b2 showed less systemic reactogenicity than did people vaccinated with BNT162b1. BNT162b2 was therefore selected to go forward to an ongoing phase II/III large-scale clinical trial6.
So what do the data tell us about whether the vaccine generates immunity to COVID-19, and about the correlates of immune protection — the quality and quantity of vaccine-induced antibody and T-cell responses elicited? The results are encouraging but inconclusive. The presence of neutralizing antibodies is correlated with protection from SARS-CoV-2 infection in monkeys7–9, and there are anecdotal reports for humans that are consistent with this10. However, a definitive interpretation of such data is complicated by the lack of standardized tests for assessing T-cell and neutralizing-antibody responses. Approaches to tackle this shortcoming are already being developed, for example by the SARS-CoV-2 Neutralization Assay Concordance Survey (go.nature.com/3iqh0jp), and the results should help to provide a way of comparing different vaccine candidates.
COVID-19 poses a riddle for the immune system
Taken together, the early clinical data for the Pfizer/BioNTech vaccine candidate hold promise, but many questions remain for this and other mRNA vaccines that target SARS-CoV-2. For example, what is the optimal dose, and what would be the best timing for a booster vaccination? How long does the vaccine-induced immune response last? Is the vaccine safe and effective in people with underlying health conditions, or those of minority-racial and -ethnic backgrounds, who are disproportionately affected by COVID-19? Whether the vaccine is safe in children should also be tested. In addition, there are logistical hurdles to consider when distributing and administering a vaccine that requires transport and storage at −80 °C. Above all, it needs to be established that the vaccine-elicited immune response prevents infection and disease.
Data to come from the ongoing large-scale phase II/III clinical trial — revealing efficacy and longer-term safety profiles — will be crucial for answering some of the remaining questions. This is especially important for pioneering RNA-based vaccines, such as BNT162b1 and BNT162b2, that lack the extensive safety record of vaccine candidates developed using a conventional approach.
The good news is that the final hurdle on the way to the finishing line — the completion of a properly controlled phase III clinical trial — is in sight. Ideally, this process will not be jeopardized by a premature rush, through an Emergency Use Authorization by the US Food and Drug Administration or other international regulators, to get a vaccine into use in the clinic before the trial has generated sufficient safety and efficacy information. As in any hurdle race, skill, speed and judgement are all needed to successfully and safely cross the finishing line.
 Read the paper: Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults
Read the paper: Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults
 Read the paper: COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses
Read the paper: COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses
 Read the paper: SARS-CoV-2 vaccines in development
Read the paper: SARS-CoV-2 vaccines in development
 COVID-19 poses a riddle for the immune system
COVID-19 poses a riddle for the immune system
 Going back in time for an antibody to fight COVID-19
Going back in time for an antibody to fight COVID-19